
Trinucleotide Repeat Disorders
When the cause of a disease can be traced to having too many copies of a certain nucleotide triplet in the DNA, the disease is said to be a trinucleotide repeat disorder. Today, there are 14 documented trinucleotide repeat disorders that affect human beings**. Huntington’s Disease is part of this group.
Some of these 14 trinucleotide repeat disorders are more alike than others. While the symptoms and the affected body parts vary by disease, scientists consider two illnesses to be similar if they share the same repeated codon as their cause. Six of the 14 trinucleotide repeat disorders have little or no apparent similarity to each other, or to the 8 remaining diseases. These 6 are described in brief at the end of this section. The 8 remaining disorders, one of which is Huntington’s Disease, all share the same repeated codon as their cause: CAG. Since CAG codes for an amino acid called glutamine, these 8 trinucleotide repeat disorders are collectively known as polyglutamine diseases (“poly” being the Greek word for “many”). (For background information on codons and amino acids click here.)
Polyglutamine diseases have much in common: Each of them is characterized by a progressive degeneration of nerve cells in certain parts of the body (for background info on nerve cells, click here.) In each disease, this degeneration first disrupts the function of certain group(s) of nerve cells. After 10-20 years, many of the affected nerve cells die. The major symptoms of these diseases are similar to one another and they usually affect people around the same time, in mid-life (although childhood cases have also been reported, as in the case of juvenile HD).
It deserves to be reiterated that while the polyglutamine diseases are similar to each other, they are not identical. Although they share the same repeated codon (CAG), the repeats for the different polyglutamine diseases occur on different chromosomes, and thus on entirely different segments of DNA. (For more info on chromosomes, click here.) Despite this fact, scientists are excited about research in any of the polyglutamine diseases because finding a way to stop the CAG repeat from occurring in one disease may help lead to a cure for the other 7 as well. While this is by no means a certainty, the possibility offers wonderful incentive to be persistent in research; eight for the price of one would certainly be a great deal!
Below you will find detailed descriptions for each of the polyglutamine disease, as well as a general description of all the non-polyglutamine diseases.
**Although only 14 trinucleotide repeat disorders are well-documented in medicine, genetic analysis has led researchers to believe that others exist as well. These disorders are even less common than the well-documented disorders and so have been more difficult to study, which leaves much of their story untold. They will be omitted here.
Polyglutamine Diseases:^
DRPLA (Dentatorubropallidoluysian Atrophy)^
Like other trinucleotide repeat disorders, DRPLA (Dentatorubropallidoluysian Atrophy) affects both the mind and body. It is characterized by abrupt muscle jerking, involuntary movements, and eventual dementia. Although these symptoms are common in the men and women of all ages who have DRPLA, young people with the disease may also be affected by progressive intellectual decline.
The Gene:
The gene involved in DRPLA lies on Chromosome 12 and is also named “DRPLA”. Typically, in asymptomatic individuals there are between 6 and 35 copies of CAG in the DRPLA allele. In a person with the disease, however, the allele has anywhere between 49 and 88 copies. At present, not enough data exist to fully understand the effect that alleles with between 35 and 49 copies of CAG will have on individuals. To learn more about alleles and more specifically, HD alleles, click here.
The Protein:
The protein product of the DRPLA gene is called atrophin-1. Although scientists are not sure about its function, the leading theory is that atrophin-1 is involved in the pathway that helps insulin take effect in the body’s cells. Since insulin helps determine how cells utilize their energy, it is essential that this pathway work smoothly so that cells can function efficiently. If there is a kink in the plan, it could spell disaster for an affected nerve cell.
How the Symptoms Come About:

The nerve cells affected in DRPLA lie in many different parts of the brain. Understanding the functions of these different parts allows us to get a better understanding of why the symptoms of DRPLA are what they are: Take first the striatum and the globus pallidus. Together, these very important regions of the brain are collectively known as the basal ganglia. The basal ganglia are important because they help plan movements and thus have a large effect on motor control. Working with other parts of the brain such as the red nucleus and the dentate nucleus (which are also damaged in people with DRPLA), the basal ganglia help to regulate each and every movement we make. When neurons in this area are damaged due to DRPLA, it’s no wonder that muscle jerks and involuntary movements become common. (For a more detailed description of the basal ganglia – written in regards to Huntington’s Disease – click here). (See Figure F-1.)

The same can be said for damage to the cerebellum, which also occurs in people with DRPLA. The cerebellum is the region of the brain where learned movements are stored. When damage occurs here, movements that were once smooth and refined become more jerky and rough since they must be constantly relearned. (See Figure F-2.)

The cerebral cortex also has a large effect on movement, particularly through the parts of it called the motor cortex and somatosensory cortex. Thus, the cerebral cortex is also involved in the motor symptoms of DRPLA. However, the tasks of the cerebral cortex reach far beyond motor control. Consider the many amazing capabilities we humans have: keen senses, the ability to speak and understand language, and the fact that we can create and use such things as logic and reason. All of these characteristics stem from functions of the cerebral cortex. Thus, when damage occurs to specific parts of the cerebral cortex, the tasks that these parts work to accomplish may become less refined. This loss of refinement may explain why people with DRPLA experience dementia when the nerve cells in the cerebral cortex are damaged. It may also partly explain the general intellectual decline in juvenile cases of DRPLA. (See Figure F-3).
Huntington’s Disease (HD)^
For an introduction to HD, click here.
SBMA (Spinobulbar Muscular Atrophy)^

SBMA (Spinobulbar Muscular Atrophy) occurs predominantly in males and is characterized by weakness and atrophy of the proximal muscles. Difficulties with swallowing and articulating speech are also common symptoms of SBMA. As the first word of its name implies, the disease mainly affects the spinal cord (“spino-“) and a part of the brain called the bulbar region (“-bulbar”). (See Figure F-4.)
The Gene:
The gene involved in SBMA is called the Androgen Receptor (AR) gene. It is located on the X chromosome, which is one of the so-called sex chromosomes. (Unlike most of our chromosomes, the sex chromosomes differ between males and females and this is why SBMA occurs predominantly in males. To learn more about chromosomes, click here.) Typically, in asymptomatic individuals there are between 9 and 36 copies of CAG in the AR allele. In a person with the disease, however, the allele has anywhere between 38 and 62 copies. At present, not enough data exist to fully understand the effect that alleles with 37 copies of CAG will have on individuals.
The Protein:
Hearing the oft-repeated statement “DNA codes for proteins” might lead one to believe that it is as simple as show me DNA and poof!, you have a protein. Actually, the process is much more complex than that. In fact, the full sequence of events is broken up into several parts, one of which is called transcription. The Androgen Receptor gene codes for a protein of the same name, the Androgen Receptor (also abbreviated “AR”). Because the AR protein is a key player in transcription, it is aptly titled a transcription factor.
In its normal state, then, the AR protein helps cells carry out the instructions contained within DNA. (For more in-depth discussion of DNA and the genetic code, click here.) However, in people with SBMA, the protein has extra glutamines, resulting from the extra CAGs in the AR gene. Although scientists do not yet have a definitive explanation as to why the extra glutamines cause degeneration of the neuron, it seems likely that the extra glutamines create an altered form of the AR protein that does not perform its actions in the same way as the normal AR. This mechanism for degeneration of the neuron is much like the one for Huntington’s Disease, as illustrated in Figure A-3.
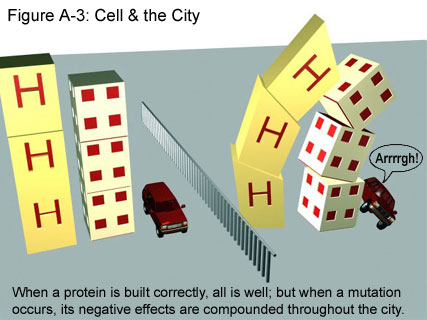{kind=link}

Another theory suggests that the degeneration of the nerve cell is a result of neuronal inclusions (NIs). This theory, too, has its equivalent in the study of Huntington’s Disease. (Click here for more about nerve cell death in HD). According to the theory, the extra glutamines in the protein have a way of attracting other proteins to group together with the AR. This aggregation of proteins causes clumps, or inclusions, which may be solely responsible for damage to the nerve cell. More research in this area is necessary to find out definitively if the NIs are the true cause of damage. (See Figure F-5.)
How the Symptoms Come About:

Whatever the mechanism, once the nerve cells become damaged, the symptoms of SBMA begin to appear. As mentioned above, one of the main areas of the body that SBMA affects is the spinal cord. More precisely, it affects the parts of the cord known as the anterior horn and the dorsal root ganglion. The dorsal root ganglion is a group of nerve cell bodies that pass sensory information to other spinal cord nerve cells and on to the brain for analysis. The anterior horn is a region of the spinal cord that contains cell bodies of motor neurons, which put the brain’s decisions (based on the sensory info) into action. These two regions of the spinal cord are thus essential for control of fine muscle movements. When the dorsal root ganglion is damaged, the brain cannot receive proper input and thus cannot plan a movement of the muscle. When the anterior horn is damaged, the brain’s planned movement cannot be carried out. Thus, if either of these regions is not functioning correctly, then the muscles are not able to carry out the same motions that they had always done before. This inability to perform normal motions is why muscle weakness and atrophy are so common in SBMA. (See Figure F-6.)

But the spinal cord is not the only body part affected by SBMA; the bulbar region of the brain is harmed as well. The bulbar region is composed of the cerebellum, the medulla and the pons. (For a tour of brain structures, including these three, click here.) An extension of the spinal cord at the base of the brain, the medulla and pons are responsible for some of the functions that keep us alive. Functions that we usually never think about, like breathing, blood circulation, and simple actions like swallowing are all in large part controlled by the medulla and pons. More complex functions, however, require use of the cerebellum. The cerebellum is where our learned movements are stored—it helps refine a great deal of motor activities, from throwing a baseball to speaking. Given the roles of the medulla, pons, and cerebellum, it’s no wonder why damage to these areas can cause difficulty swallowing and articulating speech, two more symptoms of SBMA. (See Figure F-7.)
SCA1 (Spinocerebellar Ataxia Type 1)^
SCA1 (Spinocerebellar Ataxia Type 1) is one of many closely related disorders collectively known as spinocerebellar ataxias (SCAs). Like all of the SCAs, SCA1 is characterized by atrophy of the cerebellum, a phenomenon that plays a role in the major symptoms of the disorder like loss of coordination and difficulty in articulating speech. Another common symptom of the disorder is decreased sensation in the limbs.
The Gene:
The gene involved in SCA1 lies on Chromosome 6 and is also called SCA1. Typically, in asymptomatic individuals there are between 6 and 44 copies of CAG in the SCA1 allele. In alleles with more than 20 copies (but still less than 44), the codon CAT interrupts the string of CAGs 1-4 times in a way that adds stability to the CAG chain. In a person with the disease, however, these stabilizing CATs are not present and the allele has anywhere between 39 and 81 copies of CAG. Thus, especially in the 39-44 CAG repeat range (where one may or may not be at risk for the disease), the CATs are very important—their existence can make the difference between having the illness and not.
The Protein:
The protein product of the SCA1 gene is called ataxin-1. Many studies of ataxin-1 have led scientists to believe that its major function may be to facilitate the maneuvering of nerve cell connections to allow learning. However, it is important to note that the symptoms of SCA1 are not directly caused by the loss of normal ataxin-1 function. Instead, it is believed that the cause of disease lies in the interaction between ataxin-1 and another protein called LANP. Scientists believe that LANP has a major effect on cell communication, which is needed for the survival of a nerve cell. When the ataxin-1 is altered, its interaction with LANP is also altered. The ataxin-1 is said to “sequester” the LANP and thus interfere with its normal activity. After a time, the sequestering of LANP appears to cause degeneration of the nerve cell.
How the Symptoms Come About:
To best explain how the symptoms of SCA1 come about, it is helpful to have an understanding of the cerebellum. (For more on the cerebellum, click here.)
Add to the equation a loss of pyramidal nerve cells (cells of a different pathway that are also involved in performing highly-skilled motions) and one can see why SCA1 can have such a large effect on one’s ability to perform movements.
The decreased sensation in the limbs of people with SCA1 is known as peripheral neuropathy. This condition comes about when the nerve cells that pass information from the limbs to the spinal cord (and on up to the brain) are damaged. Since they cannot do their jobs to maximum effectiveness, some of the sensory information is lost and this results in the decreased sensation.
SCA2 (Spinocerebellar Ataxia Type 2)^
SCA2 (Spinocerebellar Ataxia Type 2) is characterized by a general slowing of some of the body’s normal processes. In addition to the loss of coordination that is common to all SCAs, people with SCA2 often develop slow or nonexistent reflexes and tend to shift the focus of their eyes from one point to another in a very deliberate manner. Partial paralysis of the eyes has even been described in some cases.
The Gene:
The gene involved in SCA2 lies on Chromosome 12 and is also named SCA2. Typically, in asymptomatic individuals there are between 14 and 31 copies of CAG in the SCA2 allele. In a person with the disease, however, the allele has anywhere between 36 and 64 copies. Individuals with between 31 and 36 copies of CAG may or may not develop the symptoms of the disease (individual results vary).
The Protein:

The protein product of SCA2 is called ataxin-2. So far, although the exact function of this protein is unknown, scientists believe that it may be involved in aiding protein-protein interaction within the cell. This would make it something of a “mediator” of communications within the cell. If this theory is correct, then when the protein is in its altered form in people with SCA2, it cannot do the same mediation that the normal form does. This loss of normal function means that essential protein-protein interactions cannot be as efficient as they were with the normal ataxin-2 involved. The end result is that the health of the cell is compromised. (See Figure F-9.)
How the Symptoms Come About:

The mechanism for the loss of coordination experienced in SCA2, due primarily to damage to the cerebellum, is more-or-less the same as the mechanism described for SCA1. (Read more about the cerebellum by clicking here.) The symptoms involving the eyes, however, result from SCA2’s effect on a different part of the brain. This region is called the midbrain. The primary function of the midbrain is to control the movement of the eyes. When neurons in this area are damaged, the eye’s movements become slower than normal and even partial eye paralysis can occur. Both of these phenomena are symptoms of SCA2. (See Figure F-10.)

The effect of SCA2 on the reflexes is explained by the damage it inflicts on the granule cells. A granule cell is a specific type of nerve cell that forwards a great deal of information on to the cerebellum. Much of this information involves the positions and movements of the limbs, as well as what parts of the skin are being stimulated at any given time. In terms of reflexes, all of this information is very important. As an example, suppose that someone is burned by a hot plate: The person must know not only what body part this sensation is coming from, but also where this part is located in space and what direction to move it in order to stop the pain. If this information is slow in getting to the brain, it can delay the reflex that is needed to deal with the pain. This slower flow of information occurs when the granule cells are damaged, causing people with SCA2 to develop slower reflexes. (See Figure F-11.)
SCA3 (Spinocerebellar Ataxia Type 3 or Machado-Joseph Disease)^
SCA3 (Spinocerebellar Ataxia Type 3) is also known as Machado-Joseph Disease. In addition to the loss of coordination that is common to all SCAs, the most common symptoms of SCA3 include bulging eyes, small contractions of the facial muscles, and general rigidity.
The Gene:
The gene involved in SCA3 lies on Chromosome 14 and is also named SCA3 (although the name “MJD1” is sometimes used instead). Typically, in asymptomatic individuals there are between 12 and 43 copies of CAG in the SCA3 allele. In a person with the disease, however, the allele has anywhere between 56 and 86 copies. At present, not enough data exist to fully understand the effect that alleles with between 43 and 55 copies of CAG will have on individuals.
The Protein:

The protein product of SCA3 is called ataxin-3. Although scientists do not know the exact function of the protein, they do know that it normally resides in the cytoplasm of the cell. In people who have SCA3, however, ataxin-3 is known to aggregate in the nucleus. Researchers suspect that this change of place may be key in understanding the initiation of the disease. (See Figure F-12.)
How the Symptoms Come About:
Of all the polyglutamine disorders, SCA3 is perhaps the most perplexing with regard to the relationship between the affected brain regions and the symptoms of the disease. Damage commonly occurs in the cerebellum, basal ganglia, brain stem, and spinal cord. While damage to these areas commonly affects a wide range of movements, it does not seem to explain why such things as bulging eyes and general rigidity would occur. Hopefully, more research in this area will soon uncover the mystery.
SCA6 (Spinocerebellar Ataxia Type 6)^
SCA6 (Spinocerebellar Ataxia Type 6) is probably the simplest of all the SCAs in terms of its symptoms: People with SCA6 predominantly experience random episodes of ataxia or slowly progressing ataxia.
The Gene:
The gene involved in SCA6 lies on Chromosome 19 and is also named SCA6. Typically, in asymptomatic individuals there are between 4 and 18 copies of CAG in the SCA6 allele. In a person with the disease, however, the allele has anywhere between 21 and 33 copies. This is the smallest number of trinucleotide repeats known to cause disease. At present, not enough data exist to fully understand the effect that alleles with 19 copies of CAG will have on individuals. Individuals with 20 copies of CAG may or may not be at risk of developing SCA6. To learn more about alleles and more specifically, HD alleles, click here.
The Protein:

Instead of its own separate protein product, the SCA6 gene codes for a subunit of the calcium channels that exist in all nerve cells. This subunit, called Alpha-1A, creates a pore in the membrane of the nerve cell, allowing calcium to enter the cell and have an excitatory effect. The excited cell can then process the inputs it has received (due to calcium’s effect) and decide whether or not it should relay this information on to other nerve cells. In this way, Alpha-1A appears to play a significant role in nerve cell communication. (For more information about how nerve cells communicate, click here). In their altered form, however, Alpha-1A subunits tend to leave the membrane and aggregate in the cytoplasm inside the cell, where they clump together and do not perform their normal duties. This movement from the membrane hinders the nerve cell’s ability to receive and process messages from other nerve cells. Since communication is essential to the survival of nerve cells, the clumping of the altered Alpha-1A subunits in the cytoplasm may play a significant role in nerve cell degeneration. (See Figure F-13.)
How the Symptoms Come About:

In SCA6, the areas most affected by nerve cell damage are the cerebellum and the Purkinje cells. Given their roles in refining motions (as mentioned in the discussion of the cerebellum), one can see how damage to these areas esults in loss of coordination. Also contributing to the symptoms is degeneration of the granule cells and the nerve cells of the inferior olive. Since these structures are involved in the input of information to the cerebellum – and likewise the Purkinje cells are involved in its output – we can see that both input and output are quite important in creating smooth, precise motions. At any given time, some nerve cells may be less affected by SCA6 than others, and this may account for the random episodes of ataxia: one group of cells may be affected one day, and another group a different day. (See Figure F-14.)
SCA7 (Spinocerebellar Ataxia Type 7)^
SCA7 (Spinocerebellar Ataxia Type 7) is the last of the SCAs to fall under the category of polyglutamine diseases. Like the other SCAs, the most common symptom of SCA7 is loss of coordination. In addition to this, people with SCA7 often have difficulties with vision.
The Gene:
The gene involved in SCA7 lies on Chromosome 3 and is also named SCA7. Typically, in asymptomatic individuals there are between 4 and 19 copies of CAG in the SCA7 allele. In a person with the disease, however, the allele has anywhere between 37 and 306 copies. At present, not enough data exist to fully understand the effect that alleles with between 19 and 29 copies of CAG will have on individuals. Individuals with 30-36 copies of CAG are considered to be in the intermediate zone; they may or may not develop the symptoms of SCA7. If they do develop symptoms, the symptoms are likely to be milder and to appear later in life than they would for people with 37 or more copies of CAG.
The Protein:
The protein product of the SCA7 gene is called ataxin-7. Currently, the normal function of this protein is unknown. Scientists suspect that when ataxin-7 proteins are altered, they tend to clump together in the nucleus, producing what are called neuronal inclusions, or NIs (NIs have also been found in certain nerve cells of people with SBMA, HD, and some other SCAs). These inclusions have been associated with degeneration of the nerve cell, but whether or not they are in fact the direct cause of degeneration is yet to be determined. (See Figure F-5.)
How the Symptoms Come About:
The loss of coordination that people with SCA7 experience results from damage to the cerebellum. This mechanism is more-or-less the same as that of SCA1. (For a more detailed explanation of this mechanism, click here.)
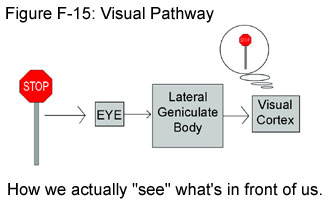
The effect that SCA7 has on one’s vision is a little more complicated because vision is a process that involves many players. Contrary to popular belief, humans do not literally “see” with their eyes. Instead, the eyes are simply the first stop on a pathway for visual information that will eventually lead to the processing of this information in the brain. After light from an image comes into the eye, the information it contains is encoded into nerve impulses by the retina. (For a discussion of nerve impulses, click here.) These impulses are then sent down the optic tract to a part of the brain called the lateral geniculate body. Here the information undergoes something like a preliminary inspection, which involves a categorization of the data. The newly categorized info is then sent on to the visual cortex, which is part of the cerebral cortex of the brain. It is in the cerebral cortex where the brain assembles a processed image and we actually “see” what is in front of us. To see an image clearly and accurately, then, all pieces in this visual puzzle must be in good working order. In SCA7, however, there is noticeable damage to all parts of the visual pathway. While this by no means implies that people with SCA7 go blind, some problems with vision are likely to occur. (See Figure F-15.)
Non-Polyglutamine Diseases^
As noted in the introduction to this chapter, polyglutamine diseases are only a subset of the trinucleotide repeat disorders. As of this writing (summer 2001), researchers have identified six non-polyglutamine diseases that also fall under the category of trinucleotide repeat disorders. Because each disease involves a unique repeated codon, the six non-polyglutamine diseases show relatively little resemblance to one another. More importantly, none of them appear to have any strong similarity to Huntington’s Disease or the other polyglutamine diseases. For this reason, we provide only brief descriptions of these non-polyglutamine disorders. The descriptions follow below.
Fragile X Syndrome^
Fragile X Syndrome (often abbreviated “FRAXA”) is a disorder involving the CGG codon (contrast this with the CAG codon involved in the polyglutamine diseases). The affected gene is called FMR1 and it lies on the X chromosome (hence the name “Fragile X Syndrome”). In asymptomatic individuals, the FMR1 allele has between 6 and 53 CGG repeats. In people with the disorder, the FMR1 allele has over 230 repeats. At present, not enough data exist to fully understand the effect that alleles with between 53 and 230 copies of CGG will have on individuals. Common symptoms of FRAXA include mental retardation, long and prominent ears and jaws, stereotypic hand movements (like flapping and biting one’s hands), hyperactivity, and others. The disease typically affects males.
Fragile XE Mental Retardation^
Fragile XE Mental Retardation (often abbreviated “FRAXE”) is a disorder involving the GCC codon. The affected gene is called FMR2 and, like the gene causing Fragile X Syndrome, FMR2 lies on the X chromosome. In asymptomatic individuals, the FMR2 allele has between 6 and 35 copies of GCC. In people with the disorder, however, the allele has over 200 copies of GCC. At present, not enough data exist to fully understand the effect that alleles with between 35 and 200 copies of GCC will have on individuals. Common symptoms of FRAXE include mild mental retardation, learning deficits, and possible developmental delays.
Friedreich’s Ataxia^
Friedreich’s Ataxia (often abbreviated “FRDA”) is a disorder involving the GAA codon. The affected gene is called X25 (also known as “frataxin”). In asymptomatic individuals, the frataxin allele has between 7 and 34 GAA repeats. In people with the disorder, the allele has 100 or more repeats. At present, not enough data exist to fully understand the effect that alleles with between 34 and 100 copies of GAA will have on individuals. There are many common symptoms of FRDA, some of which include slurred speech, heart disease, and diminished reflexes of the tendons. The name “ataxia” describes a loss of coordination, and this is typical in the limbs and trunk of those who have FRDA. The typical age of onset for this disorder is early childhood.
Myotonic Dystrophy^
Myotonic Dystrophy (often abbreviated “DM”, not “MD”) is a disorder involving the CTG codon. The affected gene is called DMPK. In asymptomatic individuals, the DMPK allele has between 5 and 37 CTG repeats. In people with the disorder, the allele has at least 50 repeats in adult-onset cases, and can go up to several thousand in congenital cases. At present, not enough data exist to fully understand the effect that alleles with between 37 and 50 copies of CTG will have on individuals. Common symptoms of adult-onset DM include muscle weakness and degeneration, while such symptoms as kidney failure, facial dysmorphology, heart problems, premature balding, cataracts, and, in males, atrophy of the testicles are less common. The congenital form of DM is the most severe and its symptoms include diminished muscle tone, problems with respiration at birth, and developmental abnormalities. The term “myotonic” comes from “myotonia”—a condition characterized by frequent muscle spasms. Obviously, myotonia is quite common in DM.
Spinocerebellar Ataxia Type 8^
Like Myotonic Dystrophy, SCA8 (Spinocerebellar Ataxia Type 8) is a disorder involving the CTG codon. The affected gene is also called SCA8. Asymptomatic individuals possess between 16 and 37 repeats of CTG in the SCA8 allele, while people with the disorder have between 110 and 250 repeats. At present, not enough data exist to fully understand the effect that alleles with between 37 and 110 copies of CTG will have on individuals. SCA8 is a slowly progressive disorder and its symptoms include decreased sense of vibration, sharp reflexes, and atrophy of the cerebellum, which has a large amount of control over the body’s learned movements. (For a more detailed description of the cerebellum, click here.)
Spinocerebellar Ataxia Type 12^
Much like the polyglutamine diseases discussed above, SCA12 (Spinocerebellar Ataxia Type 12) is a disorder involving the CAG codon. But unlike the polyglutamine diseases, which have CAG repeats that occur in what is known as the “translated region” of DNA, the CAG repeats in SCA12 occur in what is called an “untranslated region” of DNA. In what basically amounts to an exception to the normal rule, the chemical information of an untranslated region of DNA is not used as instructions for making proteins. None of the codons in the untranslated region of DNA produce any amino acids at all (a realization that has prompted some scientists to refer to the untranslated region as “junk DNA”). This exception means that the CAG codons of SCA12 actually do not produce the amino acid called glutamine. Because of this fact, SCA12 is not considered a polyglutamine disorder.
The affected gene in SCA12 is also called SCA12. Asymptomatic individuals possess between 7 and 28 repeats of CAG in the SCA12 allele, while people with the disorder have between 66 and 78 repeats. At present, not enough data exist to fully understand the effect that alleles with between 28 and 66 copies of CAG will have on individuals. SCA12 is the most recent addition to the group of spinocerebellar ataxias. Since there are relatively few cases to date, the full effects of the disorder are not yet fully known. Given that it is a spinocerebellar ataxia, however, it is likely that some of the general symptoms include slurred speech and loss of coordination of some parts of the body.
For further reading^
- Cummings, C. J. and Zoghbi, H.Y. “Trinucleotide Repeats: Mechanisms and Pathophysiology.” Annu. Rev. Genomics Hum. Genet. 2000. 1:281-328.
A fairly technical paper explaining the symptoms of each trinucleotide repeat disorder, as well as a breakdown of the codon involved and the amount of repeats in people with and without the disease (as of the publishing, however, updated and slightly different data regarding the numbers are available; see next entry in bibliography). Also discussed are theories regarding the function of the altered proteins. - GeneClinics. Online.
An in-depth site with very recent information about all of the SCAs (and DRPLA). A wonderful resource to find out more about each disorder. (Look up the any of the SCA’s by using the search feature.) - Online Mendelian Inheritance in Man (OMIM). Online.
A compilation of abstracts from a multitude of different studies on HD. From case studies regarding inheritance to new methods of diagnosing HD, this is an excellent site for all the various types of HD research going on today. (Look up any of the trinucleotide repeat disorders using the search feature.) - Silverthorn, Dee Unglaub. “Human Physiology.” Upper Saddle River, NJ: Prentice Hall, 2001. pp. 256-263, 396.
Written for college students, this textbook has excellent explanations of all aspects of human physiology, as well as wonderful pictures to increase one’s understanding. The pages noted are excellent in teaching the functions of various parts of the nervous system. - Thompson, Richard F. “The Brain.” New York: Worth Publishers, 2000. pp. 11-16, 296-303, 308-309, 451.
An introduction to neuroscience. Very clearly explains the functions of the various parts of the nervous system. Also gives insight into current research going on in neuroscience.
-M. Stenerson, 9-25-01