
The Inheritance of Huntington's Disease (Text and Audio)
Click on the link below to hear an audio recording of this article:
The Inheritance of Huntington’s Disease
Although records of symptoms have been traced as far back as the Middle Ages, it was not until the late 1800s that physician George Huntington first documented the hereditary nature of the disease that bears his name. It was the late onset and hereditary character that distinguished HD from other diseases with similar symptoms. With so many recent breakthroughs in human genome research, we now know quite a bit about the genetic basis of Huntington’s disease. Having a working familiarity with the basic genetics of HD is key to understanding the inheritance and expression of the disease.
What is a gene? Which parts of the genetic code correspond to genes?^
The material behind genetic inheritance is a surprisingly simple chemical substance called deoxyribonucleic acid (DNA). Each molecule of DNA is a long, continuous chain of smaller molecules called “bases” that are strung one after the other. The four different bases (abbreviated A, T, C, and G) can be arranged in many different ways. It is the sequential order of these bases that provides the chemical information or “instructions” for inheritance. The DNA ladder can be very, very long, twisted and coiled again around proteins into the shape of chromosomes that sit inside each cell’s nucleus. A chromosome is, in fact, one very long “super coiled” molecule of DNA. (For more on the chemical information in DNA, click here.)

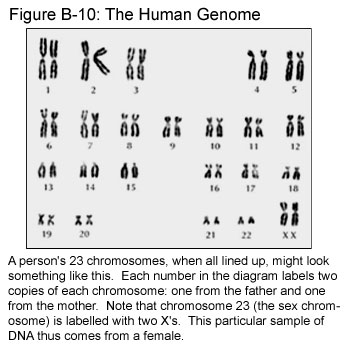
Some regions of DNA contain instructions for making a specific functional product, such as a protein. These regions of functional DNA are called genes. Every long molecule of DNA has some segments that are genes and some that are not. Together, they make up the structures called chromosomes that are found in cell nuclei. (See Figure C-1.) The non-gene segments are sometimes half-jokingly called “junk in the genome” – we still have little idea of what, if anything, these sequences do! Fortunately, though, the genes are always found in the same place on a particular chromosome. The gene responsible for causing HD, for example, is always located on chromosome 4. (Humans have 23 pairs of chromosomes, each of which has been assigned a conventional number. Click here for a look at all 23 pairs.) When scientists say that they have “located” a gene for a disease, they usually mean that they have found a region on one of the chromosomes that codes for a protein that somehow contributes to causing the disease.
{kind=link}
What are alleles? How many alleles can there be for a gene and how many copies does each individual have?^
Suppose we choose a particular chromosome from two different people and examine the DNA from the same spot on both chromosomes. We will find that the pattern of the bases (A’s, C’s, T’s, and G’s) is similar, but it is often not exactly the same, even if the region is a protein-coding gene. How can a gene code for a product if the pattern is not the same in every person? The answer is that there can be many different versions or variants of a given gene. These different versions of the gene are called alleles. Different alleles of a gene code for the same trait, but they may manifest themselves in different ways. The gene for eye color contains the instructions governing eye pigment, for example, but the specific color is determined by the particular alleles one has. Everyone has the same number of chromosomes and genes, but each person’s genetic code has a unique combination of alleles. This potential for variation explains why we all have similar genomes, yet we still have people of different heights, weights, and faces.
The way in which the Huntington gene varies among individuals is by the number of repeated C-A-G codons it contains. In other words, different alleles of the Huntington gene contain different numbers of CAG codons. It is important to understand that everyone has the Huntington gene, but individuals with Huntington’s disease have a many-CAG version of the gene, one that does not function normally. “Having the HD allele” is somewhat loose terminology, but it is used often and usually implies “having one of the multiple-CAG alleles on the Huntington gene that causes HD.” Within this site, the allele on the Huntington gene with the normal number of CAG repeats (the allele that does not result in HD) is referred to as the non-HD allele. The allele of the Huntington gene with the extra CAG repeats (the allele that does result in HD) is described as the HD allele.
How do genes determine physical traits? What does it mean to say that the HD gene is dominant?^
Since we inherit one complete set of DNA from each parent, chromosomes occur in pairs called “homologues.” (Click here for a picture of homologues.) Hence, a gene that is found on a given chromosome actually has a partner on its matching, or homologous, chromosome. This means that a person actually has two copies of every gene, one allele on each of two homologous chromosomes. This feature raises some important questions about how alleles interact and relate to each other.
{kind=link}

Alleles can be thought of as having different “strengths.” If two different alleles are present together, the “stronger” one will influence the trait under consideration. This phenomenon is called dominance. A dominant allele influences the resulting trait whether an individual has one or two copies of that allele. In contrast, in order for recessive alleles to be expressed, an individual must always have two copies of the “weaker” allele. (See Table C-1.)
HD is called a dominant trait because individuals with just one copy of the HD allele typically develop HD symptoms. The HD allele (with many CAG repeats) is dominant over the non-HD allele (with few CAG repeats). Again, an individual need have only one copy of the HD allele to inherit the disease. There is also no exact cut-off point for when the number of CAG repeats is considered “abnormal.” (Click here for a table of repeats and their effects.) Occasionally, an individual will have an allele whose CAG codon count falls within a small “gray area” for which the repeat number is slightly higher than normal, but not quite “abnormal.” This version of the allele has a medium strength. Its dominance is said to be “incomplete,” and individuals with this allele may or may not develop the disease.
{kind=link}
How are alleles inherited? What are the chances of inheriting the gene?^
Since the Huntington gene is not on a sex-determining chromosome, the disease is not sex-linked. In other words, the inheritance and development of Huntington’s disease are not related to an individual’s sex. This means that males and females have an equal chance of inheriting the disease. Males and females with the disease are also equally likely to pass it on to their children.
Every person inherits two copies of the Huntington gene, one from each parent. Likewise, every person will also pass one of these two copies to each child. The chances of giving either of these two alleles to a child are equal (50/50). A person with Huntington’s disease has one non-HD allele and one HD allele. Hence, there is a 50% chance that the non-HD allele will be passed on and a 50% chance that the HD allele will be passed on. This means that each child of an individual with HD has a 50% chance of getting the HD allele. Individuals with a chance of inheriting the disease are sometimes described as “at-risk.” At-risk individuals have the option of undergoing genetic testing, which shows whether their “50% risk” of developing HD is in reality nearly 0% or nearly 100%. (For more information about genetic testing for HD, click here.)
Individuals without any copies of the HD allele do not have HD, and these individuals are very unlikely to pass HD on to their children. They have two non-HD alleles, and the child will always receive one of these two alleles. The only exception is in the case of a new mutation, a heritable change in a person’s DNA. Very rarely a mutation will occur so that a child’s allele differs from that of the parent from whom it was inherited. Only very rarely, therefore, does an individual without an HD allele have a child with HD. For the same reason, Huntington’s disease does not typically “skip” generations. That is, we do not observe families in which a grandparent and grandchild have HD but the child’s parents do not. If such a pattern were observed, it would be most likely that one of the parents has an HD allele, but has not yet developed symptoms of the disease.
Are my children at risk?^
Let’s switch gears and think about this question from the perspective of the child of a person with HD. The child inherits one allele from each parent. The parent without HD has two non-HD alleles, so the allele from this parent will be non-HD regardless of which one is inherited. The parent with HD has one non-HD allele and HD allele. There is an equal probability of passing either of these alleles to the child. Thus the child has a 50% chance of getting the non-HD allele and a 50% chance of getting the HD allele. Since the chance of getting an HD allele is one in two, the child has a 50% chance overall of inheriting the disease. (See Figure C-2.)

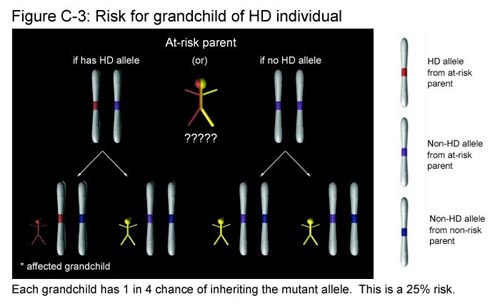
What if you discover that you are the grandchild of a person with HD, and your parent who is at risk chooses not to be tested? Recall that you have two copies of the Huntington gene. One copy (allele) will come from the parent who is not at risk. This copy will always be non-HD and does not affect your chances of getting the disease. The second copy comes from your at-risk parent. Since this parent is the child of an individual with HD, he or she has an equal chance of having either two non-HD alleles or one non-HD and one HD allele. (We found this out in Figure C-2.) In the first case, this parent has two non-HD alleles and you will not inherit the disease regardless of which of the two non-HD alleles you get. In the second case, the parent has one HD allele and one non-HD allele. Here, you will inherit the disease if you get the HD allele, but not if you get the non-HD allele. Out of the four possible outcomes, exactly one results in your having a copy of the HD allele. This represents a 25% chance that you have inherited the disease. (See Figure C-3.)
There has never been any history of HD in my family…how could the HD allele just suddenly appear?^
Remember that HD alleles are distinguished by the number of CAG codons they contain. The number of repeats is not fixed between generations, and it is possible that the number of repeats changes when cells divide. Usually this change results in a larger number of repeats, although occasionally the number of repeats decreases. No one is sure exactly what causes the number of repeats to multiply, but there is some evidence that codon numbers expand as a result of DNA copying inaccuracies during sperm formation. When DNA is copied, it is reproduced in small sections that are strung together later to make the long, continuous strands of DNA in chromosomes. There is some speculation that codon repeats could expand if these pieces are not hooked together correctly. (For more information about mutations, click here.)
A person who has a “normal allele” with a borderline number of repeats (typically between 36 and 39 copies of CAG) may produce a sperm or egg cell that contains an allele with a few additional codons. On rare occasions, these extra codons may be just enough to cause the child inheriting the allele to have an abnormal repeat number. One researcher speculated that about 10% of HD cases are caused by such changes in repeat number. Although in most cases the HD allele is not inherited in this way, this possibility explains how HD sometimes seems to just “appear” in a family.
If I have the gene, what does that mean? Will it definitely express itself, and if so, when will this happen?^
Historically, an individual known to have an HD allele almost always developed symptoms of the disease, unless he or she died of some other cause prior to onset of symptoms. Individual cases have varied greatly in severity and in rate of progression for reasons that are still not yet fully understood. As a very general rule, the typical age of onset for adult-onset HD is between the ages of 30 and 50. In most instances, individuals live with the disease for 10 to 25 years. Several studies indicate that the number of CAG codons plays a role in how soon symptoms appear. The general trend appears to be “the greater the number of repeats, the earlier the onset of the disease” although there is considerable variation. There is also evidence to suggest that the average age of onset is later for individuals who inherited the HD allele from their mothers than for individuals who inherited the allele from their fathers. It follows then that, as a general rule, onset occurs earlier when the HD allele is inherited via one’s father.
About 10 percent of all HD cases are classified as the juvenile form, which has an age of onset between infancy and 20 years. The juvenile form generally occurs when the number of CAG codons is especially large (on the order of 55 and above). Individuals with juvenile HD have different symptoms, such as rigidity, seizures, and dementia, than do individuals with adult HD. In addition, the progression of juvenile HD is usually much more rapid. (Click here for more about juvenile Huntington’s disease.)
Figure C-4 shows the correlation between increasing number of CAG repeats, from 39 to 50, and decreasing age of onset. The shaded bars show the median age of onset for individuals with a given number of CAG repeats. The exact numbers used for this graph are shown in Table C-2. Table C-2 also shows, in its third column, a way to represent the range in age of onset that occurs at each number of repeats. The figures in the third column correspond to 85% confidence intervals (C.I.) around the average age of onset. A confidence interval means that for a given number of repeats, we can be 85% sure that the actual age of onset lies within the given age range. This range is represented below in Figure C-4 by yellow bars. Please note that these range figures must be interpreted carefully: they specifically do not imply that an individual who remains symptom-free throughout a given range is exempt from HD. Any particular individual may very well develop symptoms at either an earlier or later age than those shown in the table.


For further reading^
- “Huntington’s Disease: A Brief Explanation” International Huntington Association. Online.
- “Huntington’s Disease” Online Mendelian Inheritance in Man. Online.
- “GuideLines for Genetic Testing for Huntington’s Disease.” Huntington’s Disease Society of America. Online.
-A. Hsu, updated 7-1-04
Podcast: Play in new window | Download