In Huntington’s disease (HD), an abnormal increase in the number of CAG repeats in the mutant Huntington gene corresponds to a long tract of glutamine amino acids in the huntingtin protein (Htt). This excessively long glutamine tract is sticky and leads to the formation of protein aggregates in brain cells. Whether these aggregates are toxic only upon formation or are formed as a cellular defense mechanism against free toxic mutant huntingtin protein (mHtt) is still hotly debated (more information about huntingtin protein aggregation can be found here); however, what is certain is that these protein aggregates are directly linked to neuron dysfunction and death. Moreover, a larger number of CAG repeats is clearly correlated with earlier disease onset and increased severity.
Although these findings suggest that HD is solely a result of the CAG expansion and subsequent protein aggregation, several observations indicate that other factors are also at play. For example, patients with the same number of CAG repeats may exhibit different disease patterns. In addition, there are genes other than the Huntington gene that also contain a CAG repeat region. Some of these genes may have a long region of upwards of 50 CAG repeats, but still produce functional non-harmful proteins (recall that HD can occur in patients with as few as 36 CAG repeats in the Huntington gene). Evidently, there are other cellular mechanisms, though not well understood, that govern how the expanded number of CAG repeats translates into the toxic behavior of mHtt and aggregates seen in the specific case of HD.
Recently, researchers have found increasing evidence that the cellular mechanisms contributing to neurodegenerative diseases resulting from protein aggregates involve disruptions to the quality control machinery in the cell responsible for identifying and eliminating these toxic proteins. In particular, a protein known as TRiC (TCP-1 ring complex) has been found to be involved in controlling whether proteins with a long glutamine tract like Htt will fold and aggregate. This article will first review how certain classes of proteins can control protein folding, then go over the case of TRiC and its role in Htt folding, and finally discuss how this knowledge might be helpful for researching HD therapeutics.
Protein Folding and Molecular Chaperones^
To understand how TRiC is involved in huntingtin (Htt) protein aggregation, it is useful to first review the players that make proteins in the cell. First, mRNA instructions are made based on the DNA blueprint in the nucleus, or the control center, of the cell. These instructions are then sent out into the cytoplasm of the cell and are read by structures known as ribosomes, the actual molecular machines that make proteins. Proteins are synthesized as long chains of amino acid building blocks, which are assembled one by one as the ribosome reads the mRNA instructions. For a more detailed breakdown of the protein synthesis process, go here.
However, proteins are not functional as a linear chain; they need to take on a certain shape or conformation in order to exert their proper functions. To assist in this process known as protein folding, proteins known as molecular chaperones bind to chains of amino acids that are newly made by ribosomes. Molecular chaperones, as their name suggests, are present during the protein-making process to ensure that newly made proteins behave normally and appropriately for their intended function. They do this by binding to the amino acid chain, thereby stabilizing it and facilitating proper folding. Another function that chaperones perform is to prevent folding errors and inappropriate aggregation, by recognizing incorrectly folded proteins and targeting them for repair or elimination. More information about molecular chaperones and proper protein folding can be found here.
Proteins folding incorrectly can lead to protein aggregation and disease. Moreover, there is a natural decline in a cell’s ability to continually make and fold protein correctly as one ages. As a result, in the study of HD, many scientists have focused their attention to the study of molecular chaperones and their role in responding to protein aggregation. TRiC is a molecular chaperone that has recently been found to be directly involved in regulating aggregation seen in proteins containing a long tract of glutamine amino acids, such as Htt.
What is TRiC and how is it involved in protein folding?^
TCP-1 ring complex, or TRiC, is a type of molecular chaperone that is composed of 8 different protein subunits, called CCT1, CCT2, all the way to CCT8 (CCT = chaperonin containing TCP-1). Together, these CCT subunits form a structure resembling two donuts stacked on top of each other. Like other molecular chaperones, TRiC’s primary role in the cell is to act as a protein-folding machine: they bind to a specific set of newly made proteins and fold them into shapes that allow them to function correctly. This process occurs first with the unfolded protein entering and binding into the center of one of the rings in TRiC. TRiC then closes up the ring with the protein inside. During this time when the protein is enveloped in TRiC, protein folding occurs. After the protein is folded, TRiC can open up its ring again and eject the folded protein. In addition to huntingtin (Htt), some other proteins that are known to be regulated by TRiC include actin and tubulin, which are very important proteins that maintain the structural framework of the cell.

In order to identify molecular chaperones that are involved in proper folding of proteins with glutamine repeats, researchers looked at a roundworm often used in lab research. This 2006 study examined which proteins are important for minimizing aggregation of proteins with a polyglutamine expansion (“poly” means “many”, so “polyglutamine” means many glutamines). The researchers achieved this by using a roundworm that was genetically engineered to produce yellow fluorescent protein containing a polyglutamine expansion of 40 glutamine repeats. Then, they suppressed each gene in the roundworm one by one and looked for changes in the polyglutamine expanded fluorescent protein. The logic behind this experiment is that if a gene is important for preventing proteins with polyglutamine expansions, like Htt, from aggregating, then blocking that gene would increase the amount of protein aggregation. As you might have expected, one of the genes that the scientists identified through this experiment is the one that encodes the molecular chaperone TRiC.
Although roundworm models are easy to manipulate in the lab, it is much more relevant to study Htt aggregation in human cells. Consistent with the previous result, it has also been found that blocking TRiC in human cells will result in increased aggregation of proteins with an expanded glutamine repeat region.
How does TRiC actually suppress huntingtin aggregation?^
So far, we’ve discussed how TRiC is involved in folding newly formed proteins, but how does TRiC prevent already formed mutant huntingtin (mHtt) from aggregating? Studies in this area of research first found that TRiC is able to directly bind to the Htt protein. This result aligns with what we previously knew about TRiC – that it acts as a molecular chaperone by assisting in protein folding. In particular, scientists have found, through several different experiments, that CCT1, one of the eight subunits forming TRiC, is especially important for binding to Htt. First, an in vivo study of the brain tissue of a transgenic mouse expressing mHtt revealed that TRiC and, specifically, CCT1 directly interacts with the Htt protein. This is very strong evidence indicating that TRiC regulates Htt activity in the brain in some way. Furthermore, by suppressing the CCT1 subunit alone, researchers still observe the same increase in Htt aggregation as when TRiC is suppressed. Even more significantly, expressing more CCT1 in cells can actually decrease the amount of Htt aggregation and toxicity in human cell culture models. All of these experiments suggest that TRiC can somehow control Htt aggregation through the CCT1 subunit.


Recent studies conducted between 2009 and 2013 from Judith Frydman’s lab at Stanford have shed light on how TRiC might control Htt aggregation through the CCT1 subunit. First, researchers from the lab found that CCT1 binds to the Htt protein on a region of 17 amino acids, called N17, right next to the tract of glutamine repeats. This N17 region is significant because deleting this region in an Htt protein with 51 glutamine repeats appears to slow down the toxic protein aggregation process normally observed. In other words, the N17 region right next to the glutamine tract plays a role in speeding up the process of Htt protein aggregation. Scientists also found that the N17 region of one Htt protein can bind to the N17 region and the glutamine tract of other Htt proteins. Based on this evidence, they hypothesize that perhaps the N17 region of the Htt protein acts as a sticky end that other Htt proteins can attach onto and gradually form aggregates.
TRiC is another protein that can bind to the N17 region of Htt through its CCT1 subunit. Using advanced imaging techniques, the Stanford researchers found that when Htt and TRiC were put together, TRiC can either encapsulate single Htt proteins or small Htt aggregates in its ring chamber. It can also attach onto the tips of larger Htt aggregate fibers. From this experiment, the researchers concluded that TRiC can inhibit Htt aggregation perhaps by blocking the N17 region of Htt protein from endlessly sticking to other Htt proteins to form large, fibrous protein aggregates associated with neuron death. In the presence of TRiC, Htt will be maintained as a small single protein or small protein aggregates that are not toxic to the cell.
How can these findings about TRiC translate to an HD therapeutic?^
Research about TRiC is still in its preliminary stages, since much of the mechanisms behind TRiC’s ability to suppress huntingtin (Htt) aggregation remains to be understood. However, the fact that we do know TRiC and specifically its CCT1 subunit are potent inhibitors of aggregation is useful for researchers who are thinking of ways to stop the formation of dangerous aggregates that are toxic to brain cells.
A study published in 2012 by Sontag et al. took this knowledge and asked the following question: because cells that express more of the CCT1 subunit had less Htt protein aggregation, can this CCT1 subunit be applied as a drug and be directly delivered to brain cells? In order to answer this question, the researchers used cell models of HD and first showed that a modified fragment of CCT1 can penetrate the cell membrane and enter the cell to exert its effects. With regards to therapeutic benefits, the researchers observed that when higher doses of CCT1 were applied to rat cells engineered to express mHtt, there was a corresponding decrease in the amount of visible Htt aggregates formed. The scientists followed up with another experiment where they applied CCT1 to cells of HD mice that were removed from the striatum, the brain area most affected in HD. They obtained perhaps an even more encouraging result: the cells receiving CCT1 treatment exhibited a higher survival rate compared to cells without treatment. This suggests that CCT1 may exert therapeutic benefits for cells expressing mHtt beyond just suppressing protein aggregation.
Conclusion^
The above research is simply a small-scale experiment that demonstrates the potential that this line of research can have for HD therapeutics. Evidently, there is a still a lot that we don’t know about how molecular chaperones like TRiC can suppress huntingtin (Htt) aggregation. The experiments up to this point have all been performed with cells in laboratory settings, and these results will have to be verified in subsequent animal studies before any conclusions can be made about their actual therapeutic benefit. However, studying TRiC has emphasized the importance of looking beyond the infamous glutamine repeats in Htt and looking at regions such as N17 to understand the mechanism of Htt aggregation and toxicity. Furthermore, the fact that direct protein delivery to the brain is already being done in mice for Parkinson’s disease research could lead the way for testing CCT1 in HD mouse models as well.
Further Reading^
Meyer AS, Gillespie JR, Walther D, Millet IS, Doniach S, Frydman J. (2003) Closing the Folding Chamber of the Eukaryotic Chaperonin Requires the Transition State of ATP Hydrolysis. Cell. 113(3): 369-381.
Behrands C, Langer CA, Boteva R, Böttcher UM, Stemp MJ, Schaffer G, Rao BR, Giese A, Kretzschmar H, Siegers K, Hartl FU. (2006). Chaperonin TRiC Promotes the Assembly of polyQ Expansion Proteins into Nontoxic Oligomers. Mol Cell. 23(6): 887-897.
Tam S, Geller R, Spiess C, Frydman J. (2006). The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 8(10): 1155-1162.
Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, Frydman J. (2009) The Chaperonin TRiC Blocks a Huntingtin Sequence Element that promotes the Conformational Switch to Aggregation. Nat. Struct. Mol. Biol. 16(12): 1279-1285.
Shen K, Frydman J. (2013). The interplay between the chaperonin TRiC and N-terminal region of Huntingtin mediates Huntington’s Disease aggregation and pathogenesis. Protein Quality Control in Neurodegenerative Diseases. Eds. Morimoto RI, Christen Y. 121-132.
Shahmoradian SH, Galaz-Montoya JG, Schmid MF, Cong Y, Ma B, Spiess C, Frydman J, Ludtke SJ, Chiu W. (2013). TRiC’s tricks inhibit huntingtin aggregation. eLife. 2:e00710.
Sontag EM, Joachimiak LA, Tan Z, Tomlinson A, Housman DE, Glabes CG, Potkin SG, Frydman J, Thompson LM. (2012). Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant Huntingtin cellular phenotypes. Proc. Natl. Acad. Sci. USA. 110(8): 3077-3082.
– J. Choi, 02-03-14
More
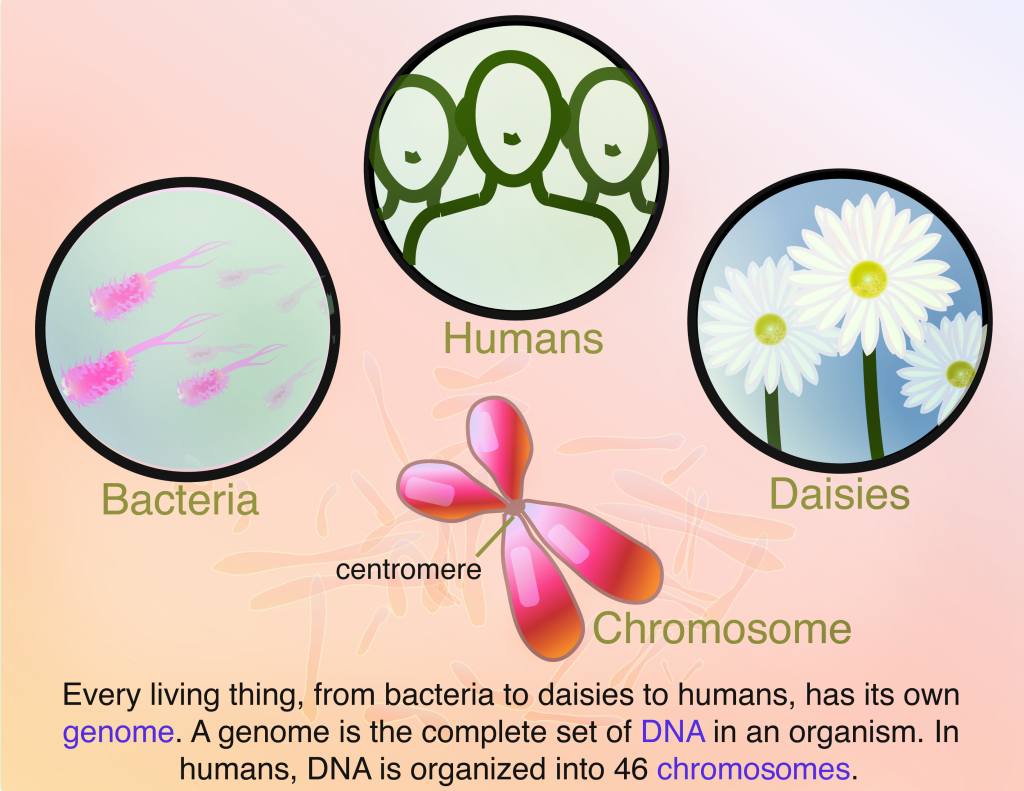
Although the pathology of Huntington’s disease (HD) is still not completely understood, we know that HD is a genetic disorder where the root cause of every HD case is a longer-than-normal series of three repeated DNA base pairs, CAG, in the HD gene. A DNA sequence provides the instructions for the cell to make mRNA (messenger RNA), which in turn contains the instructions for making a protein – the building blocks and machines of cells. The process of using the information contained in genetic material (DNA and RNA) to form protein is called gene expression. Genetic changes in the HD gene sequence are thus propagated into the mRNA sequence and result in production of a mutated version of the huntingtin protein that ultimately results in degeneration of brain cells.
In order to develop a therapy that prevents the production of the mutant huntingtin protein, many scientists are currently using a technology known as gene silencing. This approach creates molecules that directly target and bind to the mRNA copies that contain the instructions for producing huntingtin protein. These molecules then use the cell’s own molecular machinery to destroy the problematic mRNA instructions (for more information about gene silencing, click here).
Gene silencing is a very promising approach, and many scientists are focusing their efforts on conducting studies and clinical trials to assess the feasibility and efficacy of gene silencing treatments for HD and other diseases. However, it has recently become possible to go one step further in manipulating gene expression. Instead of targeting the intermediate mRNA copies as in gene silencing, some scientists are pursuing strategies that will directly modify the DNA blueprint in cells and ultimately living organisms. This article will discuss this nascent technology known as genome editing and its potential as an HD therapeutic.
What is Genome Editing?
Just as editing text involves adding, removing, or replacing words, genome editing is an approach in which the genome sequence is directly changed by adding, replacing, or removing DNA bases. However, the genome is relatively resistant to change. DNA in the body is not only responsible for encoding all of the necessary functions within a cell, but it is also crucial to determining differences between individuals. If DNA could be easily altered, many essential cell functions would be disrupted in undesirable ways. To deter any changes from being inadvertently made to DNA, cells have inherent mechanisms to proofread and repair their genetic code. These repair mechanisms and the overall stability of DNA is what makes genome editing such a novel approach and so difficult to achieve.
Remarkably, researchers have been able to take advantage of the cell’s DNA repair mechanisms to achieve genome editing. To accomplish this, scientists can use artificially engineered enzymes called nucleases to cleave DNA strands. In effect, these nucleases act as molecular scissors that form a break in the DNA double-stranded helix. Once a break is introduced in the DNA, the cell will detect a problem in its genetic code and quickly activate its repair machinery.
There are two major methods by which a cell can repair a break in its DNA. First, the cell can employ various enzymes to directly join the two ends of the DNA break back together. This process, known as nonhomologous end-joining, is very error-prone and often results in mutations – such as small insertions or deletions of nucleotides – in the resulting DNA strand. These small mutations can be neutral, but they can also render the entire gene in that location nonfunctional, achieving the disruption or knockout of the gene.
Second, the cell can also repair a DNA break by using another DNA sequence as a template. In genome editing applications, a DNA sequence can be designed to be inserted along with a nuclease, such that when a cut is made in the DNA, the cell’s own repair mechanisms can use the DNA sequence supplied to replace an existing DNA sequence as it repairs the break. This method allows scientists to directly change genetic information in cells by introducing a correct version of a DNA sequence to replace an unwanted mutation.
How can a specific gene be edited?
By using a cell’s own repair mechanisms, scientists can disrupt or correct a mutation by genome editing, and both approaches could prove useful in the context of HD treatment. However, there must be a way to direct a nuclease to the desired location where a DNA break is to be introduced. To address this issue, many different types of nucleases have been developed. All nucleases consist of 2 components – the nuclease itself that is responsible for DNA cleavage and a secondary component responsible for recognizing a specific DNA sequence. There are three main classes of nucleases engineered for genomic editing purposes:
Zinc finger nucleases (ZFNs)
ZFNs consist of a nuclease component linked to a DNA-binding component derived from an array of zinc finger proteins. Each zinc finger protein can bind three nucleotides, so combinations of zinc fingers linked together can be designed to recognize specific genomic sequences.
Transcription activator-like effector nucleases (TALENs)
TALENs are very similar to ZFNs in that they also have a nuclease domain linked to a DNA recognition domain. The difference lies in the fact that in TALENs, the DNA recognition domain is a series of amino acid repeats. Each repeat corresponds to a single nucleotide base (A, G, C, or T), and TALENs can be designed to have different combinations of repeats to recognize specific genomic sequences.
CRISPR/Cas System
The CRISPR/Cas system employs a nuclease called Cas9 to introduce a DNA double strand break. Unlike ZFNs or TALENs, this approach does not use a protein-based DNA recognition domain. In order to guide the Cas9 nuclease to a specific DNA binding site, an RNA sequence is designed to precisely bind to a complementary DNA sequence, allowing for the Cas9 nuclease to make a cut.
Meganucleases
Unlike ZFNs or TALENs, each meganuclease has a long recognition sequence that allows them to make DNA breaks at specific sites. However, these long recognition sequences are naturally defined and cannot be engineered, thus meganucleases can only be used for some target genetic sequences.
Can genome editing be used therapeutically?
Just like gene silencing, genome editing is already being used by scientists as one of their many tools to develop cell and animal models for studying different diseases. Is there a possibility that genome editing can be used in humans to cure genetic diseases like HD? Preliminary research suggests that genome editing may be a promising therapeutic approach, but more work is needed prior to clinical testing in humans.
Research has shown that simply delivering engineered zinc finger proteins (ZFPs) that do not have any nuclease activity is able to reduce the levels of mutant huntingtin. A study published in 2012 by a research group in Spain found that ZFPs can be designed to bind longer CAG repeats more strongly than shorter repeats, which means that these ZFPs could specifically recognize the mutant huntingtin gene with the CAG expansion. They further demonstrated that ZFPs reduced the levels of mutant huntingtin by 95% without affecting the levels of the wild-type huntingtin protein in an in vitro model of HD using mouse cells expressing a human version of the mutant HD gene. Moreover, they were able to demonstrate similar results in an HD mouse model, where ZFP treatment reduced the level of mutant huntingtin up to 60% and motor performance as measured on a rotarod was significantly improved. This study not only demonstrated that ZFPs can specifically bind to the mutant huntingtin gene, but also suggested that ZFPs can accomplish gene silencing by simply binding to the DNA and preventing the gene from being transcribed. These findings support the use of zinc finger nucleases (ZFNs), which could add to the repressive effect of ZFPs by actually disrupting or correcting the mutant gene.
ZFNs have been tested as a therapeutic approach in other diseases. For example, Sangamo Biosciences, a biopharmaceutical company, has explored the potential of ZFNs in treating hemophilia, a genetic disorder in which the ability of the blood to clot is impaired. The researchers used ZFNs to replace the mutated gene responsible for causing hemophilia with a correct gene that allows for normal function in a mouse model of hemophilia, and found that clotting times of the mice returned to normal after treatment. This study suggests that ZFNs are a viable strategy for correcting the genome in genetic diseases such as HD.
Conclusion
Many factors will still need to be considered before genome editing can be used as a viable therapeutic option for HD. Designing nucleases to be specific for one genetic sequence is a difficult and often expensive process, as it requires linking together the right combination of zinc fingers in ZFNs or the right combination of amino acid repeats in TALENs. Moreover, just like in gene silencing, DNA recognition by ZFNs or TALENs is not perfect and can result in off-target effects (binding to the wrong genetic sequence). Genome editing is still in its early stages, and it will be awhile before we will know if it can be used as a gene therapy for HD patients. But, many scientists are pursuing this new avenue of research with promising results.
Further Reading
Jasin, Maria. (1996) Genetic manipulation of genomes with rare-cutting endonucleases. Trends in Genetics. 12(6): 224-228.
de Souza, Natalie. (2012) Primer: genome editing with engineered nucleases. Nature Methods. 9: 27.
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotech. 31: 827-832.
Carroll, Jeff. (2011) Cut-and-paste DNA: fixing mutations with ‘genome editing’. HDBuzz. http://en.hdbuzz.net/038.
Sangamo Biosciences. ZFP nucleases: http://www.sangamo.com/technology/zf-nucleases. Huntington’s disease: http://www.sangamo.com/pipeline/huntingtons-disease.html.
Garriga-Canut¬¬¬ M, Agustin-Pavón C, Hermann F, Sánchez A, Dierssen M, Fillat C, Isalan M. (2012) Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc Natl Acad Sci USA. 109(45): E3136-E3145.
-J. Choi, 10-24-13
More
Contrary to what one may think, the brain is the most fat-rich organ in the body. Aside from being an efficient way to store energy from the food we eat, fat molecules, known as lipids, have many variations in their structure, allowing for a correspondingly large number of additional functions. For example, some lipids are integral structural components of the membrane that encloses all cells, while others act as hormones that serve as important chemical messengers between different parts of the body. In the brain and the rest of the central nervous system, lipids play critical roles as signaling molecules that trigger processes such as forming new neuronal connections and promoting brain repair. Moreover, lipids are a significant component of myelin, the coating around axons that insulates the transmission of electric signals between brain cells just like insulation on electrical wires. For more information about the role of fats in the nervous system and in HD, click here.
Recently, one family of lipids known as gangliosides has emerged as a potentially important player in HD progression. Specifically, researchers found lower levels of one type of ganglioside in not only different types of HD mouse models, but also cells isolated from HD patients and postmortem human HD brain samples. This article describes how gangliosides normally function in the nervous system, how ganglioside function may be disrupted by HD, and how these findings might be useful in the development of a viable HD therapeutic.
Gangliosides and Diseases of the Nervous System
Gangliosides are a family of lipids first identified in 1942 and so named because they were isolated from ganglion cells of the brain. Although gangliosides have since been found in cells throughout the body, they are most concentrated in the nervous system, where they seem to exhibit important effects in cell signaling and neuroprotection. However, their functions are not well understood.
One way of assessing the importance of a molecule in any biological system is by looking at the consequences if the molecule is no longer present. In the case of gangliosides, many characterized neurological disorders have been linked to defects in ganglioside production. For example, Guillain-Barré syndrome, an acute inflammatory disease that affects the peripheral nervous system, is caused by the production of antibodies by the immune system that specifically target gangliosides in the body. This type of autoimmune disease, wherein one’s own immune system inadvertently destroys a component of the body, damages the nerve axons that are responsible for nerve signaling and can result in patient paralysis. Moreover, a genetic mutation that impairs an enzyme important to the production of one type of ganglioside, resulting in complete loss of that ganglioside, leads to a severe infantile-onset epilepsy syndrome characterized by symptoms such as brain atrophy, seizures, and chorea, all of which are symptoms associated with juvenile HD (for more information about juvenile HD, click here). Since a consequence of the loss of gangliosides is neurodegeneration, gangliosides are thought to play important neuroprotective roles. In support of this theory, scientists who have genetically engineered mice lacking certain types of gangliosides have found that these mice exhibit severe neurodegeneration and accompanying motor defects that resemble those of HD mouse models.
On the other end of the spectrum, increased levels of gangliosides in nerve cells can result from overproduction of the lipid or problems in its degradation, leading to abnormal buildup. The genetic disorder Tay-Sachs disease, found mainly in Jewish populations, results from the harmful accumulation of gangliosides in the nerve cells of the brain and other tissues. Tay-Sachs disease is caused by a genetic mutation that impairs proper degradation of gangliosides. Buildup of the lipid causes nerve cells to become swollen, leading to deterioration of cognitive and motor skills. Improper control of ganglioside levels has also been observed in cases of Alzheimer’s disease, a neurodegenerative disease characterized by protein aggregates (for more information about Alzheimer’s disease, click here). Researchers have found that gangliosides bind with amyloid β-protein and facilitate the production of amyloid β-protein aggregates that accumulate in the brains of some Alzheimer’s disease patients. What is clear is that both the deficiency and excess of gangliosides in the nervous system result in neurodegenerative defects, suggesting that the careful maintenance of ganglioside levels is important for neuronal function.
Gangliosides and HD
Taking into account the above observations that abnormal ganglioside levels are often implicated in diseases of the nervous system, scientists began to question whether HD may also involve ganglioside irregularities. It was found in 2010 that the production of GM1, a specific type of ganglioside, appears to be impaired not only in different cell models of HD, but also in cells directly derived from human HD patients. In this study, Simonetta Sipione and her research group at the University of Alberta first used an in vitro model of HD by growing rat striatal cells that have been engineered to express mutant huntingtin. By using a protein marker that specifically identifies the GM1 ganglioside, they found that the levels of GM1 in the cells expressing mutant huntingtin were significantly lower compared to normal cells. More importantly, they observed the same results when they performed the experiment on skin cells isolated directly from human HD patients. The researchers also found that the levels of an enzyme involved in the production of GM1 were decreased, suggesting that the reason for the lowered levels of GM1 in HD is because of defects in GM1 production.
To determine whether the decreased levels of GM1 have any effects on striatal neuron survival, Sipione’s group of researchers administered GM1 to the cells that express mutant huntingtin. They found that the recovery of GM1 levels in the cells was accompanied by a drop in the number of cells undergoing apoptosis, or programmed cell death. On the other hand, when they added a molecule that lowers the amount of GM1 in normal cells, they observed an increase in the number of cells that underwent apoptosis. These two experiments suggest that GM1 may be an important protective factor in HD – the presence of GM1 may protect cells in the face of stress, but mutant huntingtin leads to decreased levels of GM1. GM1 deficiency in turn contributes to increased cellular apoptosis that corresponds to neuronal loss in HD.

In a second follow-up study, Sipione’s research group asked whether GM1 could be used as a potential therapeutic in HD mouse models, given the above observations that suggest a potential neuroprotective role. To answer this question, Sipione used a well-characterized type of HD mouse model that contains a copy of the human mutant huntingtin gene in its genome. This HD mouse mirrors many of the motor and cognitive symptoms of HD seen in human patients and provides a model of the disease that is useful for testing potential therapeutics (for more information about animal models of HD, click here). Importantly, these HD mice also exhibited low levels of GM1 compared to wild-type control mice. The researchers applied therapeutic GM1 by infusing the lipid into the mouse brains. They report that mice already beginning to exhibit HD motor symptoms before the treatment had restored normal motor control in four different test of motor function. This result is particularly remarkable because the GM1 treatment began following the appearance of symptoms in mice, yet still resulted in complete motor recovery. Post-symptomatic treatments are important when developing a human therapy because of the difficulties involved in treating an individual carrying the HD mutation throughout their lifetime.
Finally, in light of these encouraging in vitro and in vivo results, the researchers were interested in how GM1 might be causing such a drastic improvement in motor control in HD mice. The researchers found that GM1 treatment caused a change in the huntingtin protein. Specifically, they found that GM1-treated brain cells express higher levels of huntingtin protein that has been modified with phosphate tags at two specific amino acid sites within the protein. Proteins that have been tagged with phosphates, through a process known as phosphorylation, often demonstrate altered activity depending on the specific location of the tag. In the case of the mutant huntingtin protein, studies in mouse models have found that if phosphates are tagged on two specific locations on the huntingtin protein – serine 13 and serine 16 – the toxicity of the mutant huntingtin protein is significantly decreased. Therefore, the observation that GM1 causes the addition of these phosphate tags at these particular sites of mutant huntingtin raises the possibility that GM1 improves motor control in mice by making the mutant huntingtin protein less harmful to neuronal cells.
Conclusion
The current research on gangliosides, especially GM1, is very promising for the development of a potential therapeutic for HD, but there remain some challenges. The current experiments have only tested GM1 treatment in cell and mouse models of HD and the transition into a human study will not necessarily yield results that are as encouraging. However, ganglioside treatments for other neurological disorders, such as Parkinson’s disease, Alzheimer’s disease, and stroke, have already reached clinical trials. These studies have met the first criteria of clinical trials by demonstrating that long-term ganglioside treatment does not pose any safety concerns (for more information about clinical trials, click here). While it has yet to be shown if the ganglioside treatments are effective in any of the neurological disorders, these studies, if successful, could pave the way for beginning clinical trials to test the therapeutic value of ganglioside treatment for HD.
Further Reading
Posse de Chaves E, Sipione S. (2010). Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 584: 1748-1759.
Christie W. (2013). Gangliosides: Structure, Occurence, Biology, and Analysis. AOCS Lipid Library. http://lipidlibrary.aocs.org/Primer/content.cfm?ItemNumber=39329.
Yu RK, Ariga T, Yanagisawa M, Zeng G. (2008). Gangliosides in the nervous system: Biosynthesis and degradation. in Glycoscience (Fraser-Reid, B.; Tatsuka, K.; Thiem, J. ed.) Springer-Verlag. Berlin-Heiderberg, Germany. pp.1671-1695.
Yamashita T, Wu Y-P, Sandhoff R, Werth N, Mizukami H, Ellis JM, Dupree JL, Geyer R, Sandhoff K, Proia RL. (2005). Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc Natl Acad Sci USA. 102: 2725-2730.
Maglione V, Marchi P, Di Pardo A, Lingrell S, Horkey M, Tidmarsh E, Sipione S. (2010). Impaired ganglioside metabolism in Huntington’s disease and neuroprotective role of GM1. J Neurosci. 30: 4072-4080.
Di Pardo A, MaglioneV, Alpaugh M, HorkeyM, Atwal RS, Sassone J, Ciammola A, Steffan JS, Fouad K, Truant R, Sipione S. (2012). Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc Natl Acad Sci USA. 109: 3528-3533.
Carroll J. (2012) Special ‘brain fat’ injection helps HD mice. HDBuzz. Web: http://en.hdbuzz.net/072.
-J. Choi, 7-31-13
More
It has long been known that melatonin, a hormone produced in the brain, plays an important role in regulating the body’s natural sleep-wake cycle by causing drowsiness and inducing sleep. The pineal gland, a small structure located beneath the center of the brain produces and releases melatonin in response to the intensity and type of light detected by the eyes. Darkness causes increased melatonin release, while light inhibits melatonin release. What results is a daytime decline and nighttime rise in melatonin levels that mirrors waking and sleeping. Thus, melatonin plays an important role as a regulator of the biological clock. Because of this, melatonin is sometimes prescribed as a treatment for sleep disorders. Disruption of normal sleep is a common symptom in HD, and more information about sleep and HD can be found here.
Outside of sleep regulation, melatonin is also involved in many human bodily processes including learning, memory, and aging. Some of these functions are brought about by the antioxidant properties of the hormone itself, while others are attributed to melatonin binding with its receptor proteins, MT1 and MT2. There has been a great deal of interest in studying these additional benefits of melatonin, since it is already an FDA-approved drug. In terms of HD specifically, researchers have recently identified melatonin as a neuroprotective agent due to its role in inhibiting the neuronal death characteristic of HD. Since melatonin acts through so many different possible mechanisms, how exactly can melatonin produce its therapeutic effects against HD and affect disease progression?

Melatonin as an Antioxidant
Free Radical Damage in HD
In order to understand the role of antioxidants like melatonin in HD, we must first briefly review free radical damage, a phenomenon implicated in HD-associated neuron death. Free radicals are highly reactive molecules that are natural byproducts of biochemical processes in the body, but high levels of free radicals can be toxic to cells in the body because they cause oxidative damage. Neuron cells in the brain, seem to be particularly susceptible to oxidative damage. For more information about free radicals and how they damage cells, click here.
One cause of free radical excess is glutamate excitotoxicity. Glutamate is an important neurotransmitter, a chemical signal used by neurons to communicate with each other. Normally, binding of glutamate to NMDA receptors on neurons is responsible for processes such as learning and memory in the brain. Scientists believe that in HD, the mutant huntingtin protein causes problems that lead to excess binding of glutamate (for more information about NMDA receptors and glutamate toxicity, click here). One of the eventual results of this defect is the overproduction of free radicals.
Another mechanism that can lead to free radical damage in HD is dysfunction of the mitochondria, the parts of the cell that act as energy power plants. A normal byproduct of the mitochondria producing energy for the cell is the generation of free radicals. However, if mitochondria are defective they may overproduce free radicals, leading to increased damage in the cell. This is one possible explanation for the mitochondrial defects observed in some patients with HD.
Melatonin’s Antioxidative Properties
As a defense mechanism against harmful free radicals that are normally produced, the body employs molecules known as free radical scavengers, which we more commonly refer to as antioxidants. As their name implies, free radical scavengers encounter free radicals and detoxify them before they can damage cells or tissues. Melatonin, in addition to being a hormone that regulates sleep-wake cycles, is also a potent antioxidant. Its antioxidant properties go beyond free radical scavenging. There is evidence suggesting that melatonin can stabilize cell membranes and thereby increase the cell’s resistance against free radicals, that it can stimulate cellular production of other antioxidants, and that it may even play a role in directly inhibiting production of certain types of free radicals. Melatonin is also among the few antioxidants that can cross the blood-brain barrier, thus extending its protective properties to neurons in the brain.
Much research has been done to determine whether melatonin is truly effective for reducing oxidative damage. In order to mimic neuronal damage as a result of uncontrolled free radical levels in HD, scientists use chemicals such as quinolinic acid and 3-nitropropionic acid. The former is a molecule that binds to NMDA receptors which mimics glutamate, while the latter interrupts mitochondrial activity, and injection of either chemical into animal models result in oxidative damage similar to the neuropathology seen in brains of HD patients. In a 1999 study performed by Southgate et al., rat brains were treated with quinolinic acid, which caused oxidative damage to cell membranes. However, after the addition of melatonin, the amount of damage decreased significantly, suggesting that melatonin has protective antioxidative effects. In a more recent 2005 study by Nam et al., 3-nitropropionic acid was injected directly into the striatum of rats, creating neuronal lesions similar to those seen in HD. Treatment with melatonin reduced the amount of free radical damage to levels comparable to control mice that did not have a 3-nitropropionic acid injection, and significantly decreased the size of neuronal lesions in 3-nitropropionic acid injected mice. All of these studies provide evidence that melatonin has protective effects against these chemically-induced HD models through antioxidant action. However, it is important to remember that these chemical treatments do not precisely replicate HD and further research must be done to demonstrate the effects of melatonin on oxidative damage in HD.

Melatonin and Melatonin Receptor Interactions
The reasons why melatonin demonstrates neuroprotective effects are still unknown, but aside from its antioxidant properties, researchers have also shown that the interaction of the hormone with its receptors could be another way it protects the brain from damage by HD. Like all hormones, melatonin is a chemical signal, and in order to bring about responses, melatonin must bind to one of its two receptors in the body – MT1 and MT2. Since altered levels of MT1 and MT2 receptors have been observed in other neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease, it is possible that the melatonin receptors can play a role in HD as well.
In 2011, Friedlander’s research group conducted a study on the interaction between melatonin and its receptors in HD models and the therapeutic potential of melatonin treatment. Their results shed light on the means by which melatonin might protect neurons. Firstly, they found that in addition to its free radical scavenging abilities, melatonin treatment in vitro also blocked cellular pathways that lead to neuron death, or apoptosis, which some research suggests is increased by the mutant huntingtin protein in HD. More importantly, they found that this effect is achieved by melatonin binding specifically to MT1 receptor. However, in brain tissue samples of both mice carrying the mutant huntingtin gene and patients with HD, levels of MT1 receptor seemed to decline along with HD progression – samples that had more severe, late-stage HD pathology also had significantly lower levels of MT1. This fact, coupled with the earlier findings that melatonin provides cell survival signals via the MT1 receptor, suggests that this interaction is disrupted in HD and is a potential target for HD therapeutics.
The Friedlander research group subsequently tested the effect of melatonin treatment in HD mouse models. In contrast to the earlier animal models mentioned which were induced by chemicals to exhibit HD-like symptoms, they chose to employ transgenic mice that carry a version of the mutant huntingtin gene. Daily melatonin treatment showed a beneficial effect on HD mice. Not only did melatonin-treated mice retain normal movement control longer before onset of disease, but they also survived 21% longer than HD mice with no treatment. Brain tissues were also less damaged in melatonin-treated mice than in untreated mice. However, some key characteristics of HD – such as weight loss and levels of mutant huntingtin protein aggregates – remained unaffected by melatonin. Nevertheless, the results of this in vivo study was a physiological reflection of the in vitro findings that melatonin binding with MT1 may help protect neurons by inhibiting certain pathways that cause cell death.
Conclusion
Melatonin’s antioxidative properties and its interaction with MT1 receptors both seem to contribute to its protection of neurons in HD and make it a promising therapeutic candidate for the disease. Although melatonin is already approved to treat certain sleep-related conditions, research on melatonin’s effect on HD progression is still in its early stages and further work must be done to validate these results before melatonin can potentially be studied in clinical trials.
Further Reading
- Wang X, Zhu S, Pei Z, Drozda M, Stavrovskaya IG, Del Signore SJ, Cormier K, Shimony EM, Wang H, Ferrante RJ, Kristal BS, Friedlander RM (2008) Inhibitors of cytochrome c release with therapeutic potential for Huntington’s Disease. J Neuro 28: 9473-9485.
- Reiter RJ, Manchester LC, Tan D-X (2010) Neurotoxins: Free radical mechanisms and melatonin protection. Curr Neuropharmacol 8: 194-210.
- Reiter RJ, Cabrera J, Sainz RM, Mayo JC, Manchester LC, Tan D-X (1999) Melatonin as a pharmacological agent against neuronal loss in experimental models of Huntington’s disease, Alzheimer’s disease, and Parkinsonism. Annals of New York Academy of Sciences 890: 471-485.
- Southgate G, Daya S (1999) Melatonin reduces quniolinic acid-induced lipid peroxidation in rat brain homogenate. Metabolic Brain Disease 14: 165-171.
- Nam E, Lee SM, Koh SE, Joo WS, Maeng S, Im HI, Kim YS (2005) Melatonin protects against neuronal damage induced by 3-nitropropionic acid in rat striatum. Brain Research 1046: 90-96.
- Wang X, Siranni A, Pei Z, Cormier K, Smith K, Jiang J, Zhou S, Wang H, Zhao R, Yano H, Kim JE, Li W, Kristal BS, Ferrante RJ, Friedlander RM (2011) The melatonin MT1 receptor axis modulates mutant huntingtin-mediated toxicity. J Neuro 31: 14496-14507.
-J. Choi, 1-27-13
More
Huntington’s disease (HD) is a genetic disease due to the abnormal CAG expansion of the mutated huntingtin gene. The mutated gene instructs cells in the body to produce a version of the huntingtin protein that ultimately leads to neuronal damage in the brain and creates the symptoms of HD. Currently, available HD therapies can only provide relief of symptoms, but what if there were a way to eliminate the root of the problem by correcting the mutated gene or preventing the production of the “bad” huntingtin protein? We are still years away from being able to directly edit the genome in human patients, but much progress has been made in the field of gene silencing, a scientific technique which can hinder or stop the production of a protein. This article discusses the mechanism of gene silencing, its potential as an HD therapeutic, and challenges this possible therapy faces on the path to clinical implementation.
What is Gene Silencing?
As the name implies, gene silencing is a technique that aims to reduce or eliminate the production of a protein from its corresponding gene. Genes are sections of DNA that contain the instructions for making proteins. Proteins are essential molecules that perform an array of functions including signaling between cells, speeding up biochemical reactions, and providing structural support for the cell. Each gene is responsible for producing a corresponding protein in a two-step process. First, a copy of the information encoded in a gene is made in the form of messenger RNA (mRNA), a process known as transcription. This occurs in the nucleus of the cell, the cellular structure where all of the cell’s genetic material is contained. The mRNA subsequently travels out of the nucleus, and the genetic information it carries is used to produce a specific protein, a process known as translation. (For more information about proteins and how they are made, click here.)
Instead of directly editing DNA or inhibiting the transcription process, the key idea behind gene silencing is intervening in gene expression prior to translation. By designing a molecule that can specifically identify and breakdown the mRNA carrying instructions for making a certain protein, scientists have been able to effectively decrease levels of that protein. Imagine the gene silencing molecule as a censor and mRNA as messages from genes that are broadcast into proteins: the molecule will censor out a specified mRNA message, preventing the corresponding protein from being broadcast into the cell, and thus silencing the gene that is providing these instructions. The ability to significantly lower the levels of a specific protein opens up many possibilities in scientific research and drug development, since proteins are critically involved in the proper function and structure of cells.

Types of Gene Silencing Techniques
There are various gene silencing methods currently employed in research and being developed as potential disease therapeutics. Nearly all of them involve disabling the function of mRNA by preventing it from being translated into a protein. However, they differ in the design of the molecule used to disrupt mRNA and the manner of mRNA breakdown. As a result, different silencing methods have specific advantages and drawbacks. Two of the leading and most understood methods of gene silencing are RNA interference (RNAi) and antisense oligonucleotides (ASOs).
RNA Interference
In RNAi, the molecules that identify the target mRNA are called small-interfering RNAs (siRNAs). Unlike normal single-stranded RNA found in cells – such as mRNA – siRNAs are short, synthetically made double-stranded RNA molecules designed to pair with a specific mRNA strand. This association of the siRNAs with a particular target mRNA causes the breakdown of the target mRNA by recruiting other proteins that degrade the mRNA target. Because siRNAs are double-stranded, they are more stable and less susceptible to degradation than ASOs, allowing them to continue to perform their silencing function for a longer period of time in the cell. For a more detailed description of how RNAi works, click here.
Antisense Oligonucleotides
Similar to siRNAs, ASOs are engineered by scientists to associate with a target mRNA strand. The binding of the ASO to mRNA directs a protein to breakdown the mRNA. However, unlike siRNAs, ASOs are smaller, single-stranded RNA molecules. As mentioned above, single-stranded RNAs are not as stable as double-stranded ones; thus, ASOs are often chemically modified to increase their durability in a biological environment. However, their smaller size and chemical structure allow ASOs to be transported in cells and living tissues much more effectively than siRNAs. For a more detailed description of how ASOs work, click here.
Is one gene silencing method better than the other?
In terms of developing a drug therapy based on gene silencing, how do RNAi and ASOs compare to each other in effectiveness? In cell culture experiments, gene silencing is often used to intentionally decrease levels of a certain protein for research purposes. In such applications, siRNAs have sometimes been shown to produce stronger and longer lasting gene silencing than ASOs. However, when developing silencing therapeutics, the strength and duration of gene silencing needed for treatment may vary; sometimes a shorter-acting or less complete gene silencing may be required. Furthermore, when considering the efficacy of each method in live animal models, the results are not as clear-cut. For example, as mentioned earlier, ASOs can often be distributed more easily than siRNAs throughout the target tissue because of their size and structure. This observation would be expected to simplify delivery and lower costs of a therapeutic application. The fact that there is no definitive answer to which gene silencing method is more effective has resulted in continued active research and development of both areas.
Gene Silencing and HD
HD is characterized by a mutation causing excess CAG repeats in the Huntington gene and the consequent production of the mutant huntingtin protein results in disease. As such, silencing of the mutant version of the huntingtin gene is a potential therapeutic strategy for HD treatment. Indeed, HD and other related neurodegenerative diseases involving mutant CAG repeats, such as spinobulbar muscular atrophy and some types of spinocerebellar ataxias, have been at the frontier of the therapeutic development of gene silencing (for more information about trinucleotide repeat disorders, click here).
Approaches to Huntingtin Gene Silencing
Recall that everyone’s DNA is composed of two copies of a gene, called alleles, one from each parent. In the majority of individuals with HD, one copy of the gene is mutated with excess CAG repeats, while the other copy is an allele with a number of CAG repeats within the normal range. As a result, the body not only produces the mutant version of the huntingtin protein, but also makes the normal protein. When considering gene silencing as a therapeutic approach to HD, it is crucial to think about the difference between silencing huntingtin mRNA in general and selectively disrupting mRNA that encodes for the mutant, and not the normal, huntingtin protein.
The huntingtin protein has many roles in proper development. Studies in mouse models have shown that completely eliminating huntingtin protein results in mice that do not survive past the embryonic stages of development, while mice that were induced to lose huntingtin after birth experienced severe neuronal degeneration (for more information about the function of wild-type huntingtin protein, click here). Thus, it is important for scientists to develop gene silencing drugs that specifically target mutant huntingtin mRNA. This type of approach is known as allele-specific gene silencing.
While it may seem straightforward to target the excess CAG repeats to specifically decrease the levels of mutant huntingtin, it is important to remember that the molecules used for silencing are short RNA sequences, about 25 nucleotides in length. Hence, they cannot effectively distinguish between the size of the normal and the expanded CAG repeats of the huntingtin gene. This is particularly true when trying to differentiate between 30 CAGs and 40 CAGs, CAG repeat ranges that are near normal. To get around this obstacle, scientists are developing an approach that identifies single nucleotide polymorphisms (SNPs) – changes in a single nucleotide in the DNA sequence –closely linked with the mutant gene and not the normal allele. SNPs are mutations that differ by a single nucleotide (e.g. ‘A’ à ‘C’) and result in the genetic variation between individuals. What researchers have found is that many HD patients have common SNPs that are associated with the mutated huntingtin allele. Using silencing molecules to identify these SNPs provides a potential approach to allele-specific silencing of the mutant gene.
A recent study explored allele-specific silencing of the mutant huntingtin protein by targeting associated SNPs. Since not all HD patients have the same SNPs, they sought to develop a panel of ASOs to maximize the coverage of the HD population. They found that 85% of HD patients can be covered by targeting as few as three SNPs. Moreover, injecting ASOs targeting an HD-associated SNP into HD mice showed a greater than 50% decrease in mutant huntingtin protein, while normal mice receiving the same treatment showed only a 3% drop in huntingtin levels. These results indicate a relatively strong and selective silencing effect on mutant huntingtin. Although further work must be done to expand coverage of the HD population by this approach and to assess its therapeutic efficacy outside of animal models, this study represents an initial step forward toward using allele-specific gene silencing as an HD therapeutic.
Although the above results are encouraging, some researchers have suggested using nonallele-specific silencing of huntingtin protein because of the lack of a single SNP that will specifically target mutant huntingtin in all HD patients. In this approach, instead of trying to decrease levels of the mutant huntingtin only, both the normal and mutated versions of the huntingtin protein are targeted and decreased. This method is also advantageous because instead of employing different silencing molecules for different SNPs in different individuals, a single therapy can be developed for all HD patients, thus minimizing costs. Although huntingtin has important functions in the body, interestingly, a study of nonallele-specific silencing using siRNA in HD mouse models found that when both mutant huntingtin and normal huntingtin levels were decreased by 75%, the treated mice demonstrated improved motor control and increased survival compared to controls. This result suggests that nonallele-specific silencing may be a beneficial therapeutic for HD. An important caveat to consider is that many therapies that show an effect in HD mouse models may not directly translate to humans. For example, the mouse brain may be better able to tolerate a decrease in normal huntingtin than the human brain. In any case, since both allele-specific and nonallele-specific silencing methods have their pros and cons, both continue to be under investigation as potential therapeutic options.
Challenges to Gene Silencing Therapeutics
Even though gene silencing is a promising strategy for treating HD, there are still many hurdles to overcome before it can be applied in the clinic. First and foremost, gene-silencing molecules have to be effectively delivered to the relevant parts of the body, which, in the case of HD, are the afflicted areas of the brain. The blood-brain barrier prevents passage into the brain of most molecules that are injected or absorbed into the blood, making drug delivery difficult. Some methods that scientists have used in animal models include direct injection of the silencing drug or implanting pumps that infuse the molecules into the brain.
Once past the blood-brain barrier, silencing molecules have to locate neurons and other affected cells and enter these cells to silence huntingtin expression. As mentioned earlier, due to their structure, ASOs distribute and enter cells more effectively than siRNAs. To effectively deliver siRNAs into cells, scientists currently use viral-based delivery systems, which essentially take advantage of the machinery viruses use to infect our cells. One of the drawbacks of using a viral delivery mechanism is the potential for an immune response against the molecules. As an alternative to this method, Dr. Jan Nolta’s group at the University of California Davis has begun studying the possibility of using mesenchymal stem cells (MSCs) as a delivery system for siRNA (for more information about MSCs, click here). A possible advantage of using a viral or stem cell delivery system is that they might be able to become a production facility for siRNA molecules, allowing long-term therapeutic treatment of HD, a great benefit for a chronic illness. Ongoing research is currently investigating whether this theoretical possibility could become a reality.
There are other concerns associated with gene silencing therapeutics. For example, researchers have observed that high dosages of silencing molecules could have a toxic effect, highlighting the importance of finding an optimal dosage that is safe and effective. In addition, there is the possibility for ASOs and siRNAs to accidentally bind to an undesired mRNA (mRNA coding for a protein that is not huntingtin). To deal with this so-called off-target gene silencing phenomenon, researchers are studying the selectivity of how siRNAs and ASOs bind to the huntingtin mRNA, in order to better develop specific, effective, and safe HD gene silencing therapeutics.
Conclusion
Gene silencing as a therapeutic strategy is a highly active area of research and may one day yield an effective treatment for HD, since it acts by directly reducing the production of the mutant huntingtin protein. However, the technology is still in preclinical stages, and there remain many issues to address and resolve before it can be approved for clinical trials. Delivery methods, dosages, and selectivity of gene silencing drugs must be optimized to ensure safety and efficacy of treatment. Clinical trials have begun using gene silencing for therapeutic applications in other diseases. These studies will help to inform current efforts to develop gene silencing for HD treatment.
Further Reading
1. Bennett CF, Swayze EE. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol. 2010. 50:259–93.
This is a technical article published by Isis Pharmaceuticals that gives an in-depth review of the mechanisms and pharmacology involved in developing RNA-targeting gene silencing therapeutics.
2. Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL. Nonallele-specific silencing of mutant and wild-type Huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Molecular Therapy (2009) 17 6, 1053–1063.
A technical article that explains the potential benefit of nonallele-specific silencing in HD mouse models.
3. Boudreau RL, Rodriguez-Lebrón E, Davidson, BL. RNAi medicine for the brain: progresses and challenges. Hum Mol Genet. 2011 Apr 15;20(R1):R21-7.
A medium-difficulty article that discusses the development of RNAi as a therapeutic, current preclinical data, and the key challenges that remain for its clinical implementation.
4. Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM, Hayden MR. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/ allele-specific silencing of mutant huntingtin. Mol Ther. 2011 Dec;19(12):2178-85.
A technical article that explains the results of a study to develop a panel of ASOs for allele-specific gene silencing of mutant huntingtin in mouse models.
5. Dessy A, Gorman JM. The emerging therapeutic role of RNA interference in disorders of the central nervous system. Clinical Pharmacology & Therapeutics (2011) 89 3, 450–454.
A medium-difficulty article that gives a broad overview of the current status of RNAi as a developing therapy for neurodegenerative diseases.
6. Scholefield J, Wood MJ. Therapeutic gene silencing strategies for polyglutamine disorders. Trends Genet. 2010 Jan;26(1):29-38.
A technical article that reviews the mechanism of gene silencing and discusses therapeutic studies that have been done and challenges that remain to be addressed for allele-specific silencing of polyglutamine disorders.
7. Sah DWY, Aronin N. Oligonucleotide therapeutic approaches for Huntington disease. J Clin Invest. 2011;121(2):500–507.
Another technical article that explains and compares the various approaches to gene silencing therapeutics for HD.
8. Sass, Meghan; Aronin, Neil. “RNA- and DNA- Based Therapies for Huntington’s Disease.” Neurobiology of Huntington’s Disease: Applications to Drug Discovery. Ed. Donald C. Lo and Robert E. Hughes. Boca Raton: CRC Press, 2010.
A technical article that broadly covers the mechanisms, current studies, and challenges of both RNAi and ASO therapeutics for HD.
J. Choi 04.04.12
More








