
The Basic Neurobiology of Huntington’s Disease (Text and Audio)
Click on the link below to hear an audio recording of this article:
The Neurobiology of Huntington’s Disease
Now let’s look at the broader picture and ask a number of questions about the way in which the C-A-G triplet repeat in the genes of people with HD manifests itself in the symptoms of HD. We’ll focus on changes in the brain that take place with the onset of HD.
What is a nerve cell?^
Each human body contains many different kinds of cells – liver cells, kidney cells, heart cells, nerve cells, and so on. The brain and nervous system are made up of a large number of one of these cell types—the nerve cell. The nerve cell, also called a neuron, is unique in its structure and function. The figure below shows what a typical nerve cell looks like. It has a main area called the nerve cell body, where most of the component parts of the cell, called organelles, are found. Among these organelles are the nucleus, where the DNA is packaged (see Introduction to DNA and Chromosomes), and the mitochondria, which supply the cell’s energy, as well as a whole bunch of other parts the cell needs in order to function. The cell body is covered with tiny projections that receive input in the form of impulses from other nerve cells. Impulses are merely messages that are transmitted as electrical signals from one nerve cell to another. There is also a larger projection called the axon that transmits impulses from one part of the nervous system to another.

How do nerve impulses work?^

Nerve impulses are tiny electrical signals that travel through the nerve cells of the body, conveying information. Within a given nerve cell, the impulses move quickly from the receptor end of the cell which has small projections called dendrites. Dendrites, found on every nerve cell, extend out from the cell body and are responsible for receiving messages from other nerve cells. The impulses then travel down along the axon to the terminal branches. There, the impulses are carried across the gap to the next neuron by way of chemicals called neurotransmitters. These chemicals are capable of binding to receptors found in the dendrites of the next receiving nerve cell. Once a number of receptors on the receiving cell have chemicals bound to them, they trigger changes in the receiving neuron that enable it to send the electrical signal down its axon.
When that signal reaches the end of the axon, it again triggers the release of neurotransmitters that are then picked up by the next receiving nerve cell. The process repeats itself over and over in rapid succession, relaying the impulse along a sequential chain of neurons, until the impulse reaches its destination.
In short, neurotransmitters are the chemical messengers that nerve cells use to communicate. There are many different types of neurotransmitters, and each nerve cell responds differently to them. Some “excite” neighboring nerve cells, and send impulses scurrying along their axons where they release abundant neurotransmitters. Others “inhibit” adjacent neurons, and cause them in turn to release fewer neurotransmitters. The overall effect is a bit like Morse code: information is conveyed as modulated signals between neurons in the brain and nervous system.
For a more detailed explanation of the nerve impulse click here.
What parts of the brain are most affected in HD patients?^
The part of the brain most affected by HD is a group of nerve cells at the base of the brain known collectively as the basal ganglia. The basal ganglia organize muscle-driven movements of the body, or “motor movement.” The major components of the basal ganglia are the caudate and the putamen (together known as the striatum) and the globus pallidus (external and internal regions). The substantia nigra and the subthalamic nucleus are often included as part of the basal ganglia as well. Below is a figure showing where the different parts of the basal ganglia are located.
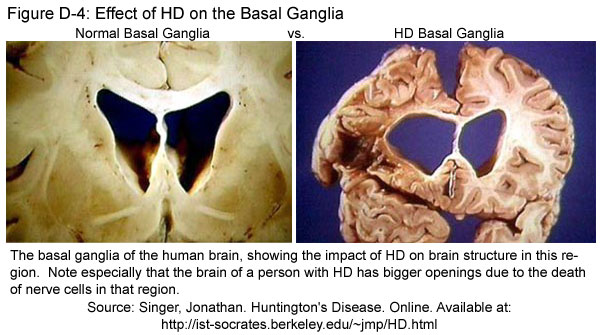
In persons with HD, the structures of the basal ganglia are a lot smaller than normal (see Figure D-4). This reduction happens because nerve cells of the striatum are the first to die as HD progresses. Although other parts of the brain are also affected during HD, the basal ganglia appear to be the most heavily damaged. Understanding the functions of the basal ganglia and the ways in which these functions are disturbed during HD can help us understand why HD affects people the way it does.
What are the functions of the basal ganglia?^
When you look at a picture of the human brain, you’ll see that it looks gray on the outside and white on the inside. The gray matter around the outside of the brain is called the cortex. Nerve cells that make up the cortex are called cortical neurons. The cortex is divided into different parts, one of them being the motor cortex. The motor cortex is responsible for the planning and execution of body movements. Scientists have found two pathways of neural connections between the motor cortex and the basal ganglia that control the coordination of such movements. As nerve cells in the striatum of basal ganglia die under the influence of HD, both of these pathways are eventually damaged.
The Direct Pathway^
The diagram below (Figure D-5) shows the direct pathway through which nerve cells communicate in sending a message from the basal ganglia to the motor cortex. As explained in more detail in the figure caption, the effect of HD on this pathway is triggered by the death of cells in the striatum. The loss of striatum cells sets off a chain of events that results in the general under-stimulation of the motor cortex. This under-stimulation from the direct pathway appears to cause the slow speed of motor movement often seen in persons with Huntington’s disease.

The Indirect Pathway^
Figure D-6 shows another way that Huntington’s disease affects the motor cortex—namely, by way of the so-called indirect pathway. Once again, the death of nerve cells in the striatum has a “ripple effect” through the rest of the pathway, resulting here in the over-stimulation of the motor cortex of the brain. This over-stimulation is believed to cause the irregular, jerky movements or chorea that have long been associated with HD. In fact, HD used to be called “Huntington’s chorea” because of these effects. However, the name was changed a few years ago because some forms of the disease, especially juvenile HD, are not associated with these irregular movements.

How can the direct and indirect pathways operate at the same time, and yet have opposing effects on the stimulation of the motor cortex? The answer seems to be that there are two different types of neurons in the striatum, one for each pathway. Although both types of neurons release the same main neurotransmitter—a chemical called GABA, which figures into some aspects of HD research today—their axons run to different dendrite targets. Research shows that the neurons of the indirect pathway are generally affected first, which explains why chorea is often seen during the start of adult forms of HD. As the disease progresses, both types of striatal neurons die off, disrupting both the indirect and direct pathways and producing an overall decrease in movement. In juvenile cases of HD, both kinds of striatal neurons degenerate from the start, which is why chorea is usually not associated with juvenile HD.
How does HD cause striatal and cortical nerve cells to die?^
Not much is known for certain about how and why specific cells die within the basal ganglia of HD patients. This section will attempt to answer some of the questions scientists are currently asking themselves.
First of all, the huntingtin protein is present in all the cells of the body, not just nerve cells. Scientists currently do not know the exact details of the function of the normal huntingtin protein in the body, but they do know that huntingtin is necessary for development and is active throughout the body. However, HD does not kill all the cells in the body; rather, it selectively kills nerve cells. The effects of HD seem to suggest that the huntingtin protein regularly interacts with proteins found only in the brain, and that the altered form of the huntingtin protein disrupts this interaction, leading to nerve cell death.
Various experiments have revealed that the huntingtin protein interacts with two proteins: huntingtin’s interactor protein (HIP-1) and huntingtin’s associated protein (HAP-1). These two proteins are present only in the brain, and this finding could explain why HD only affects the brain even though the huntingtin protein is present throughout the body. The number of C-A-G repeats in the huntington gene determines how the huntingtin protein interacts with HIP-1 and HAP-1. As repeat numbers increase, huntingtin binds less to HIP-1 and more to HAP-1. Much information about how these proteins interact and what these interactions have to do with HD has yet to be discovered. The figure below shows the proposed interaction of the altered huntingtin protein and HAP-1.

Other questions scientists are attempting to answer include: Why is the striatum predisposed to damage? Why are certain populations of striatal neurons selectively targeted during the start of HD? A couple of theories have been presented, but scientists are still working on determining the exact events involved in the progression of cell deaths caused by HD.
One theory proposes that neurons die in HD because of an over-accumulation of normal excitatory chemicals involved in nerve impulses. Excitatory chemicals are important, and they are normally present in the brain. However, if they are released in excessive amounts or if brain cells are weak, these excitatory chemicals can cause cell damage and become chemicals known as “excitotoxins.” One of the neurotransmitters released by the basal ganglia is called glutamate, which acts as an excitatory neurotransmitter in the brain. Studies show that when glutamate is injected into the basal ganglion region of brains of living rats, the rats exhibit symptoms of HD because glutamate apparently acts as an excitotoxin when there is too much of it. Scientists speculate that striatal neurons may be receiving too much glutamate from other nerve cells that release glutamate as a neurotransmitter. Other experiments have also shown that nerve cells that receive too much glutamate will die.
This first theory had to be modified when high levels of glutamate were not found in the brains of all HD patients. The modification has to do with mitochondria – a type of organelle (mentioned near the beginning of this section) that produces energy in animal cells. The mitochondria of striatal cells may be damaged with the onset of HD, leading to their increased susceptibility to even normal levels of glutamate. Scientists have since then done away with the idea that too much glutamate is being released by other cells. Rather, scientists currently believe that the damaged mitochondria of people with HD make striatal cells unable to produce as much energy as they need, which then makes the cells more susceptible to normal levels of glutamate.
Another theory to explain the death of nerve cells postulates that the cells actually kill themselves in response to chemical changes caused by HD. The theory proposes that HD triggers the early death of neurons by accelerating a normal process called apoptosis. Scientists are studying whether the presence of the altered huntingtin protein—a molecule produced within the nerve cells themselves—causes nerve cells to die prematurely. (See Basics of Huntington’s Disease.) Researchers have inserted the human HD gene into mice in order to see what happens to the nerve cells of these modified mice. They found that nerve cells of these mice contained clumps of protein material in their nuclei called neuronal inclusions (NI). They then looked at nerve cells of people with HD and saw that such clumps were also present in their nerve cells. Further studies have revealed that these clumps actually contain only a part of the altered huntingtin protein, suggesting that the altered huntingtin is, at some point, cut into fragments, some of which end up as clumps in the nucleus.
But there is a further complication: all living cells continually break down and destroy proteins as part of their normal activity, either because the proteins are misfolded, are no longer needed, or are aberrant in some way. What’s puzzling is that the altered form of huntingtin is not completely broken down by the cell. Why not, when so many other aberrant proteins are degraded all the time in animal cells? Scientists are currently looking at reasons for this anomaly. Studies are also being carried out to determine whether the NIs actually cause nerve cell death or are only by-products of some other problem.
To sum up, the neurobiological effects of HD appear to be the result of a number of different changes that ultimately go out of control. Neurobiologist Anne Young has proposed a model for how HD disrupts cell function. According to Dr. Young, the problem starts when the extended C-A-G repeats of the huntingtin gene code for an altered form of the huntingtin protein. The altered huntingtin then interacts with various proteins in nerve cells and causes the nerve cells of people with HD to become very sensitive to glutamate. This increased sensitivity leads to the activation of other proteins called caspases that cleave huntingtin to smaller fragments. The fragments then slip into the nerve cell’s nucleus and interfere with the normal production of other proteins. This interference causes cellular stress that could then lead to more huntingtin being broken up into fragments, initiating a cycle that eventually leads to the death of the nerve cell.
Researchers believe that increased understanding of how HD progresses will further the development of new pharmaceutical drugs and other therapies that will one day serve as treatments for HD. In fact, one implication of the neurobiology reviewed here is that different drugs might well be used someday to target different stages of HD progression. For example, drugs to lessen the sensitivity of nerve cells to glutamate could be administered at the start of HD. Later on, as the disease progresses, drugs that prevent the huntingtin protein from being cleaved could be given. Hopefully, with continued neurobiological research, treatments such as these will become a reality and HD will cease to be a devastating disease.
For further reading^
- Cha, Jang-Ho and Young, Anne B. Huntington’s Disease.Online.
This page contains lots of information about the HD, including its history, neurobiology, and genetic aspects. - Quarell, Oliver. Huntington’s Disease: The Facts (Oxford Medical Publications). Oxford University Press, 1999.
This book is an absolutely great resource for anyone interested in HD. It contains lots of information about the neurobiology and genetics of HD in an easy-to-understand format. It’s available at the following web sites:
- The Huntington’s Disease Society of America: www.hdsa.org
- Barnes and Nobles: www.barnesandnoble.com
- Amazon.com: www.amazon.com
-E. Tan, 8-22-01
Podcast: Play in new window | Download