
"Normal" huntingtin and Huntington’s disease
This article will seek to provide an overview of current understandings of what huntingtin protein does and why it is important.
Since the Huntington’s Study Group first identified the mutation responsible for Huntington’s disease (HD) in 1993, there have been many studies conducted seeking to understand how this defective gene causes the drastic neurodegeneration seen in individuals with HD. It is known that the expansion of CAG repeats in the Huntington gene results in a misfolded protein product commonly referred to as mutant huntingtin1,2 (For more information of how huntingtin is mutated in HD, click here). Many studies have focused on the role of mutant huntingtin in the development and progression of HD, with experimental evidence suggesting that this abnormal protein is toxic to the neurons of the striatum and other regions of the brain (For more information on how mutant huntingtin affects the brain, click here). Despite the importance of understanding how mutant huntingtin causes neurodegeneration, there is also a great deal of scientific interest in studying the function of wild-type huntingtin, the “normal” un-mutated version of the protein. Understanding how the normal protein functions may provide clues as to how the mutated version contributes to HD. But even after seventeen years of research, there remain questions about the nature, structure, and function of wild-type huntingtin.
Sequence and Structure^
Although wild-type huntingtin is expressed throughout the human body, it is most concentrated in the male testes as well as the neurons of the central nervous system (especially those in the neocortex, cerebellar cortex, striatum and hippocampus). Huntingtin is a large protein with a mass of 347 kilo-Daltons that is made up of 3,144 amino acids. Near the beginning of its amino acid chain, the protein contains a repeating region of 11 to 34 glutamine residues that are encoded from a region of CAG repeats on DNA (For more information on the mutation that gives rise to HD, click here). For more information on how proteins are made, click here). Expansions of the number of glutamines in this part of the protein, called the polyglutamine region, is known to cause HD. Although understanding this expanded region has been crucial to the study of huntingtin and HD, its relevance to the function of the entire protein is not yet well-understood.
Studies comparing the sequence of human huntingtin to that of other organisms revealed that huntingtin appears to be quite an old protein in evolution. The gene that encodes huntingtin is highly conserved among vertebrates, meaning that similar sequences are found in many different animals including mice, rats, pigs and fish. Similarities can also be found in more distant relatives such as fruit flies, although there are more significant differences in the protein’s sequence and function as compared to humans. For example, the gene that encodes for huntingtin in fruit flies does not contain a sequence of CAG repeats. This evidence hints at the possibility that when humans and fruit flies diverged along the evolutionary tree, the function of their huntingtin evolved to differ as well. Going even further back in evolution, the amoeba Dictyostelium discoideum appears to have a gene similar to the one that encodes for huntingtin, suggesting that the gene encoding for huntingtin evolved a long time ago.
Determining the present-day function of huntingtin in humans remains elusive, although progress has been made in characterizing the wild-type protein’s actions. Experiments that analyzed huntingtin’s sequence of amino acids, also known as its primary structure, have found that it does not seem to share much of its sequence with other proteins in the human body. Nevertheless, several sites of interest have been found in the proteins sequence. Researchers have determined that huntingtin contains multiple regions of so-called HEAT repeats, a sequence of ~40 amino acids named after the first four proteins in which it was discovered:Huntingtin, Elongation factor 3, a subunit of protein phosphatase 2A and the lipid kinase TOR. Although the exact function of HEAT repeats are currently unclear, studies have suggested that these domains play a role in a variety of interactions between proteins, including transportation in the cytoplasm and nucleus, microtubule dynamics and chromosome segregation. The identification of 37 potential HEAT repeats in huntingtin suggests that the normal function of huntingtin may involve some of these protein-protein interactions.
Further analyses of the sequence of huntingtin have revealed that the protein contains several other regions of interest. Huntingtin has been shown to contain a functional nuclear export signal (NES), a sequence of amino acids that allows the protein to travel out of the nucleus The nucleus of mammalian cells are separated from the cytoplasm by the nuclear envelope, a membrane consisting of two lipid layers. Small molecules can move through pores in the nuclear envelope, but large molecules do not fit through these openings. In order for large proteins such as huntingtin to travel across this membrane, they first need their NES bound by helper proteins known as exportin, which can then facilitate movement across the nuclear envelope through specialized channels. This ability of huntingtin to exit the nucleus seems to also depend on a 17 amino acid sequence near its polyglutamine region that interacts with a nuclear pore protein on the nuclear envelope. Removing this sequence causes huntingtin to accumulate in the nucleus.

The NES in human huntingtin has been shown to be highly conserved in the huntingtin of other vertebrate species, with only minor changes in the NES sequence that do not significantly change the function of this domain. Generally, the more conserved a sequence of DNA is across different species, the more important this region is likely to be to the functionality of the protein. After all, important sequences are not likely to change through evolution.
Huntingtin also contains several sites are that are known to be targeted by caspases, proteins that cleave other proteins into smaller fragments. Experiments have observed that both wild-type and mutant huntingtin are cleaved by caspases, although the mutant protein seems to be more susceptible to cleavage and its fragments are more likely to be found in the cytoplasm and nucleus. There is now strong evidence that the fragments resulting from the cleavage of mutant huntingtin are key to the progression of HD (For more information on the significance of huntingtin cleavage in HD, click here). However, the importance of cleaving in wild-type huntingtin has yet to be determined.
The primary structure of huntingtin gives some clues to its three-dimensional structure. Analyses of other proteins with HEAT repeats show that these regions form alpha-helices, coiled cylindrical structures that are hydrophobic, or repel water. For proteins with HEAT repeats, over 50% of their final structures have been shown to be made up of alpha-helices, which tend to form superhelices, helices that are themselves twisted into helices. Because of the way that they are arranged, superhelices have a core that is very hydrophobic. An experimental study that examined copies of laboratory-engineered huntingtin found that the folded protein’s structure is dominated by alpha-helices that stack to form a superhelix with a hydrophobic core. Indeed, the authors of this study suggest that the folded structure of huntingtin is a long superhelix in the shape of a solenoid.
Potential Functions^
Analyses of the sequence and structure of huntingtin have provided hints to the protein’s functions in the human body. These findings are supported by various experimental studies that directly tested for huntingtin’s function and determined that huntingtin is involved in a number of important processes at both the cellular and the organismal level. These processes include transport of molecules within a cell (intracellular transport), regulation of transcription, inhibition of programmed cell death (apoptosis), and embryonic development. These functions will be discussed in detail below.
Intracellular transport^
Huntingtin’s many roles in intracellular transport have been well-documented in scientific research (intra = “within” so intracellular literally means “within cells”). Experiments have consistently observed that huntingtin is involved in the intracellular trafficking of vesicles, spherical containers made up of lipids that transport molecules around the cell. One mechanism for this action appears to be huntingtin’s interaction with microtubules, the rod-like components of the cytoskeleton that aid in the transport of vesicles. Microtubules are made up of stacks of proteins called tubulin, which constantly add on to and fall off of the ends of these stacks to lengthen or contract the length of these structures as necessary. This dynamic growth and dispersion is necessary for many important cell processes, such as the separation of chromosomes during mitosis (For more information about the role of microtubules in dividing cells, click here). Microtubules also provide the scaffolding for specialized molecules known as the microtubule motor proteins to move across the cytoplasm. These motor proteins can be thought of as cellular deliverymen, moving along the microtubule highways to carry their vesicle cargo from one place in the cell to another.

Huntingtin has been shown to interact with a variety of different components involved in the action of microtubules, including tubulin and dynein, a microtubule motor protein that usually moves towards the center of cells. Mapping studies that examined the sequence of huntingtin found a binding site for dynein, suggesting that the two proteins can interact within cells. This finding has been corroborated by evidence that huntingtin forms complexes with dynein in cell extracts from mouse neurons. In addition to the direct action of huntingtin, its binding partners have also been proposed to affect microtubule dynamics. Huntingtin-associated protein 1 (HAP1) is one such partner that interacts with dynactin, a protein that is essential for dynein function, and kinesin, a microtubule motor protein that usually moves towards the cell periphery (the opposite direction of dynein along microtubules). Indeed, both huntingtin and HAP1 are transported in axons at a speed consistent with the hypothesis that they move along microtubules with microtubule motor proteins. In addition, huntingtin and HAP1 are capable of moving towards both the cell nucleus and the periphery, as would be expected if these proteins associate with dynein and kinesin. The direction in which huntingtin moves depends on the signals it receives from other interacting proteins. Based on observations such as these, scientists have suggested that huntingtin acts as a molecular scaffold, allowing different proteins to come together and interact.
Huntingtin’s interactions with microtubules, dynein, and other elements of intracellular transport can help explain its effect on vesicle trafficking. For example, huntingtin appears to be important in the intracellular transport of brain-derived neuronal factor (BDNF), a neurotrophic factor important for striatal cells to develop and survive (For more information on the relationship between HD and BDNF, click here). BDNF is initially produced by neurons in the cortex and substantia nigra and then moved to the striatum, the major site of neurodegeneration in HD. Like all secreted proteins, BDNF must first be synthesized within cells before being released. After BDNF is translated from its messenger RNA, it is processed and packaged into vesicles that are transported to the cell membrane along the microtubule “highways.” When these BDNF-containing vesicles reach the membrane, they accumulate in specialized compartments until they receive the signal to be released. Huntingtin appears to be crucial in the transport of BDNF from its site of production to the cell membrane. In vitro experiments in which abnormally high levels of huntingtin were expressed in neuronal cell lines found that this protein increased the speed that BDNF-containing vesicles moved along microtubules. Conversely, knocking down the huntingtin levels reduced vesicle speed. Interestingly, expressing mutant huntingtin also reduced the transport of BDNF, thereby reducing the ability of neurons to release this factor that is crucial for other neurons to survive. These results provide a compelling model for understanding one way that HD might cause neurodegeneration in the brain.
Although the transport of BDNF is a well-studied example, huntingtin has been shown to affect a broad range of the cellular machinery involved in intracellular trafficking. For example, one study found that the knockdown of huntingtin in zebrafish, a popular model organism in scientific experimentation, caused them to become iron deficient. Although zebrafish embryos with decreased levels of huntingtin could take in iron, their cells were incapable of transporting iron to where it was needed. Future studies investigating the role of huntingtin in intracellular trafficking and transport are likely to be important in understanding the molecular mechanisms that underlie HD.
Transcriptional regulation^
Huntingtin appears to help regulate transcription by acting as part of an on/off switch that tells cells when to begin expressing certain genes. One mechanism through which huntingtin regulates transcription is by binding to transcription factors, proteins that regulate the transcription of DNA to make messenger RNA. This direct interaction seems to be one mechanism by which huntingtin controls the expression of BDNF. The gene that encodes for BDNF is preceded by a regulatory region on the DNA called the repressor element 1/neuron restrictive silencing element (RE1/NRSE). This region can be recognized and bound by a transcription factor called repressor element-1/neuronal restrictive silencing transcription factor (REST/NSRF). The binding of REST/NSRF to RE1/NSRE turns off the expression of the gene that comes after this regulatory region, which in this case is BDNF. Huntingtin has been shown to directly bind to REST/NSRF, keeping it from entering the nucleus and binding to RE1/NRSE. As a result, the BDNF gene can be expressed. In HD, mutant huntingtin allows REST/NSRF to enter the nucleus and block BDNF transcription, resulting in a deficit of this critical neurotrophic factor. Interestingly, the RE1/NRSE sequence not only regulates the BDNF gene, but is also found before other genes involved in neuronal development and function. Computational analyses have suggested that the human genome contains almost 1,900 RE1/NSRE sequences, suggesting that huntingtin may play a role in the transcriptional regulation of many other genes.
Huntingtin’s role in regulating transcription is consistent with its ability to shuttle between the nucleus and the cytoplasm. Although huntingtin was once thought to be present only in the cytoplasm of neurons, both the full-length protein and its fratments have been detected in the nucleus. Since huntingtin is found in the nucleus, it must be able to cross the nuclear envelope that separates the nucleus from the cytoplasm. Most large proteins that enter the nucleus have a nuclear localization signal (NLS), which has the opposite function as NES and enables proteins to enter the nucleus (see above section for an explanation). However, analyses of huntingtin’s structure found that it does not contain a classical NLS, suggesting that the protein has an alternative means of crossing the nuclear envelope. The ability of huntingtin to both enter and exit the nucleus has led researchers to suggest that huntingtin is involved in a complex of proteins that shuttle molecules between the nucleus and the cytoplasm. Scientists hypothesize that this shuttling may be involved in regulating which transcription factors are allowed into the nucleus at certain times. However, as most studies have focused on the effects of mutant huntingtin on transcription, this role of wild-type huntingtin remains speculative.
Inhibiting apoptosis^
A role of huntingtin in neuroprotection was suggested by the finding that turning off the gene for huntingtin in adult mice caused them to develop severe neurodegeneration. The huntingtin-deficient mice exhibited a phenotype that was similar to that seen in an HD mouse model. Conversely, over-expressing huntingtin in mice protected neurons from certain kinds of neuronal damage, with higher levels of huntingtin conferring more neuroprotection. The protective qualities of huntingtin have been attributed to its ability to inhibit apoptosis, or programmed cell death. Researchers have observed that apoptosis in vertebrate neurons depends on the action of caspases, the enzymes that cleave other proteins into smaller fragments. Huntingtin has been observed in vitro to block the formation of a protein complex that activates caspases to begin the apoptotic process. Similar evidence of apoptosis was observed in the brains of adult mice and zebrafish that had huntingtin gene turned off.
Embryonic development ^
While huntingtin’s various cellular functions are usually attributed to its actions in neurons, the protein appears to also be essential for the development of embryos. Studies that have knocked out the gene for huntingtin in mice found that their embryos die within seven or eight days after fertilization. Furthermore, it is not enough for huntingtin to simply be present in the developing embryo, but it must be present in sufficient quantities. Mice that have 50% or less of the normal levels of huntingtin survive after the eighth day post-fertilization, but show significant defects in the development of their nervous system. These mice are usually able to reach adulthood, but show behavioral abnormalities, cognitive defects and neurodegeneration in certain areas of their brains. Interestingly, adding back copies of huntingtin with the expansion mutation (even those with up to 128 CAG repeats) can rescue knockout mice from an early death, suggesting that the role of huntingtin in embryo development is different from its actions later in adult life. This hypothesis is supported by evidence that humans who are homozygous for the mutant gene that causes HD (i.e. they have no “normal” copies of the gene) have no apparent birth defects.
Wild-Type Huntingtin and HD^
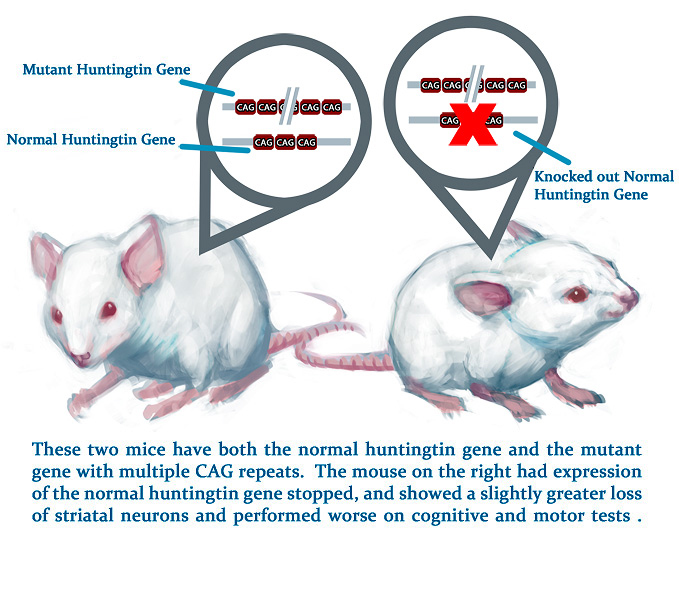
Given huntingtin’s many important functions in the adult brain, scientists have suggested that wild-type huntingtin may affect the development and progression of HD. This hypothesis is supported by studies that have observed that humans and mice that are homozygous for the CAG expansion (and thus do not produce any wild-type huntingtin) appear to have a more severe disease progression. These observations have been supported by experiments that have manipulated the levels of huntingtin in animal models of HD. One study used a strain of HD mice with a mutant huntingtin gene containing 128 CAG repeats. These mice were then mated with other mice that had their huntingtin gene knocked out producing some offspring with only mutant huntingtin and some with both mutant and wild-type huntingtin. The HD mice that did not produce wild-type huntingtin showed a slightly greater loss of striatal neurons and performed worse on cognitive and motor tests. However, these negative effects were not very severe, suggesting that the progression of neurodegeneration in these HD mice was primarily due to the presence of mutant huntingtin rather than the absence of wild-type huntingtin.
There is also evidence suggesting that over-expressing wild-type huntingtin may be beneficial in HD. Experiments examining the effects of mutant huntingtin in mice testes found that adding wild-type huntingtin reduced the extent of cell death usually seen in these HD mice. Similar results have been observed in the central nervous system of HD mice that were genetically engineered to also over-express wild-type huntingtin. However, the benefits of increased levels of wild-type huntingtin were deemed to be modest, suggesting that simply adding more of the wild-type protein will not be sufficient in treating HD.
Although there is still considerable uncertainty about the functions of wild-type huntingtin, scientists have identified several important roles that the protein plays in the cell as well as in the body as a whole. Further research into wild-type huntingtin would be valuable not only because it can give a better picture of what goes wrong in the mutant version of the protein, but may even offer clues for developing future treatments and cures.
Notes:
1) Unless otherwise specified, when this article uses the term “huntingtin” it is referring to the wild-type form. The mutated form of this protein is explicitly referred to as “mutant huntingtin.”
2) In other articles, HOPES has opted to refer to the gene encoding for huntingtin as the “Huntington gene” in order to emphasize its crucial role in Huntington’s disease. In this article alone, the gene will be referred to as the “huntingtin gene” to differentiate the wild-type huntingtin gene and the disease caused by the mutation in this gene.
Sources:^
Borrell-Pagès, M. et al. (2006). Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cellular and Molecular Life Sciences 63: 2642-2660.
In this review article, the authors summarize the functions of wild-type huntingtin and what goes wrong when it is mutated in HD. The writing is very technical.
Cattaneo, E. et al. (2005). Normal huntingtin function: An alternative approach to Huntington’s disease. Nature Reviews Neuroscience 6: 919-930.
This comprehensive review summarizes the different functions of wild-type huntingtin. While it does get technical when talking about specific experiments, the article is generally quite readable.
Caviston, J. and E. Holzbaur (2009). Huntingtin is an essential integrator of intracellular vesicular trafficking. Trends in Cell Biology 19(4): 147-155.
In this technical review article, the authors discuss the importance of huntingtin as a “scaffold protein” that is needed in the intrascellular transport of vesicles.
Colin, E. et al. (2008). Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. The EMBO Journal 27: 2124-2134.
In this very technical primary source article, the authors provide evidence that huntingtin affects the movement of vesicles in neurons by directly interacting with various components of the transport machinery.
Gauthier et al. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118: 127-138.
This study presents convincing evidence that huntingtin affects the transport of BDNF along microtubules. As a primary source article, the subject matter is very specific and the language is quite technical.
Gissi et al. (2006). Huntingtin gene evolution in Chordata and its peculiar features in the ascidian Ciona genus. BMC Genomics 7: 288.
This primary source article provides a detailed (and technical) genomic analysis of the evolution of the huntingtin gene by looking at the gene in sea squirts.
Harjes, P. and E. Wanker (2003). The hunt for huntingtin function: interaction partners tell many different stories. Trends in Biochemical Sciences 28(8): 425-433.
In this review article, the authors present the evidence suggesting that huntingtin interacts with many other proteins to execute its cellular functions. The language is very technical.
Imarisio, S. et al. (2008). Huntington’s disease: from pathology and genetics to potential therapies. Biochemical Journal 412: 191-209.
In this technical but readable article, the authors review the role of wild-type huntingtin and discusses possible molecular mechanisms for HD.
Li et al. (2006). Expression and characterization of full-length huntingtin, an elongated HEAT repeat protein. The Journal of Biological Chemistry 281: 15916-15922.
This primary source article presents evidence that huntingtin’s HEAT repeat regions causes it to fold into a structure dominated by alpha-helixes that form a superhelix.
Sandou, F. and S. Humbert (2009). Huntington’s disease: Function and dysfunction of huntingtin in axonal transport. In P. St. George-Hyslop et al. (eds.) Intracellular Traffic and Neurodegenerative Disorders. 115-123.
This chapter of a very specific and technical book provides evidence that huntingtin affects the transport of vesicles in axons.
Truant, R. et al. (2007). Nucleocytoplasmic trafficking and transcription effects of huntingtin in Huntington’s disease. Progress in Neurobiology 83(4): 211-277.
In this technical review article, the authors discuss the evidence suggesting that huntingtin is involved in regulating transcription.
Xia, J. et al. (2003). Huntingtin contains a highly conserved nuclear export signal. Human Molecular Genetics 12(12): 1393-1403.
The authors of this primary source article presents evidence for a nuclear export signal in huntingtin that is highly conserved in evolution.
Zuccato, C. et al. (2010). Molecular mechanisms and potential therapeutic targets in Huntington’s disease. Physiological Reviews 90(3): 905-981.
This recent review gives a nice overview about the molecular biology of HD, including the genetics of model organisms, the function of wild-type huntingtin, the mechanisms of neurodegeneration, and potential targets for therapies. Even with this breadth, the article is quite technical.
Yuan, J. and B. Yanker (2000). Apoptosis in the nervous system. Nature 407: 802-809.
This succinct article reviews the various causes of apoptosis in the nervous system, especially in the context of neurodegenerative disease. Although the language is very technical, the many images greatly aid in comprehension.
Y. Lu 2010