Huntington’s disease presents unique psychosocial issues due to its late onset and hereditary nature. One of the major issues of course is stress, which can come from many sources and has many effects (for general discussion of stress and HD, click here). A major source of psychosocial stress associated with HD comes from predictive testing which became available in the United States in 1993.
Extensive research has focused on the person undergoing predictive testing, with a good number of studies reporting that the tested person’s benefit from the knowledge of their genetic status outweighed their post-test psychosocial distress. However, less research has focused on the psychological impact that predictive testing may have on those at risk for HD and their partners, family and friends. This research is important because HD affects many more people than just the person who has it. Moreover, the hereditary nature of the disease can also lead to difficult questions about reproduction and about the possibility of other family members having the disease.
Fortunately, researchers are now focusing more of their attention on predictive testing and its effects on the couple relationship. In the remainder of this section, we review their key findings to date.
What percentage of couples looks favorably upon predictive testing? And what motivations drive their decisions?
In a 1989 study in Belgium, where HD predictive testing has been available since 1987, Evers-Kiebooms found that a moderate majority of people at-risk for HD and their non-carrier partners looked positively on predictive testing. Out of 349 study subjects, 66% of the at-risk adults and 74% of their partners wanted testing for the at-risk individual. The difference between these percentages can partly be explained by the difference in motivations between the at-risk person and his/her partner in approving the predictive testing. When asked why they approved, at-risk adults tended to cite worries about their futures, while their partners tended to cite worries about current and/or future children. A reason that some couples decided not to undergo predictive testing was concern about the effects of the testing on their relationship; this concern was more often a major consideration for non-carrier partners than for the at-risk adults. (For specifics on the process of HD predictive testing, please click here.
Is there a theory on how predictive testing affects couple relationships?^
Yes, a perspective called family systems theory, developed over the past few decades, has proven particularly useful in genetic counseling. This theory is especially relevant to the genetic counseling of couple relationships because its central focus is on the family rather than the individual. The family systems theory describes human behavior as a consequence of family relationship patterns, rather than individual psychology. Consequently, family systems theory can help explain the effects of predictive testing on couple relationships by analyzing how family relationship patterns can influence post-test behavior.
In a 2004 study by Richards and Williams, 43 couples were divided into two groups: those that chose to undergo predictive testing and those that chose not to. Couples in both groups answered the same questionnaire before predictive testing, then 6 months later (3 months after those tested received their test results), and again 24 months after the first questionnaire. The questionnaire consisted of 32 Dyadic Adjustment Scale questions that measured couple relationship functioning, known as a “couple score.” Those couples that received higher couple scores frequently interacted and communicated with each other, rarely disagreed with each other on significant marital issues, and settled disagreements in a way that was satisfying to both partners.
The major finding of this study was that, over the 24 month period, there was no statistically significant difference in couple scores between couples who had decided to undergo predictive testing and couples who had decided not to. The key conclusion was that predictive testing has few negative effects on couple relationships. As the authors noted, this conclusion matches the findings of several other studies (Tibben et al., 1993a; Cordori and Brandt, 1994; Quaid and Wesson, 1995; Taylor and Myers, 1997. For a look at these studies, please see “For Further Reading” at the end of this chapter).
An additional finding from the 2004 study is interesting. The couples that underwent predictive testing were categorized into couples in which the at-risk partner was a carrier and couples in which the at-risk partner was a non-carrier. Unexpectedly, the carrier couples had higher couple scores (stronger couple relationships) of statistical significance than the non-carrier couples. This suggests that, for some couples, the knowledge that their at-risk partner did not have HD had a greater negative effect on their marital relationship than the knowledge that their partner did have HD. The authors give a possible explanation: “The threat of HD may have served as a factor in the continuance of the relationship. Once this threat is removed, partners may no longer feel a duty or need to remain in the marriage to care or to be cared for.”
Another possible explanation is provided by examining family patterns via family systems theory rather than individual behavior. Family systems theory suggests that the couple relationship can be negatively affected when one or both partners have different expectations for the predictive test’s results. When the results prove to be different from expectations, conflict can arise contributing to relationship deterioration and lower couple scores. Studies by Huggins et al. and Soldan et al. have found that professional genetic counseling can benefit the couple relationship by helping partners discuss their expectations of the predictive test’s results and their coping strategies (See “Further Reading” below for links to these two studies).
What does the medical literature say about the pros and cons of predictive testing for couple relationships, especially psychosocial aspects?^
Similar to the work of Richards and Williams reviewed above, a study by Decruyenaere in 2004 also used the Dyadic Adjustment Scale to measure changes in the couple relationship for 5 years following predictive testing. But the study also collected qualitative data from separate interviews with the at-risk persons and their partners. Qualitative data are useful because they can provide more thorough explanations for trends observed in couple relationship over time. The specific couple relationship examined in the Decruyenaere study was marriage.
In this study, all at-risk persons were undergoing predictive testing, with 26 carriers and 14 of their partners, and 33 of non-carriers and 17 of their partners participating in the study. The main finding was that the majority (70%) of the tested persons did not have a change in marital status over the 5 years of the study. As for the quality of the marital relationship, half of the couples reported no change in that interval compared to the quality before the predictive testing. Out of those that did report change, non-carrier couples cited less distress and more communication. Carrier couples that experienced increased relationship quality over the five years cited more mutual support.

A conclusion that can be drawn from this study is that the test result does not by itself predict outcomes in the couple relationship; even couples with negative test results for HD may experience post-test psychosocial distress and couple relationship breakdown. The important factor for couples undergoing predictive testing is whether the test result causes role shifts that upset the balance of the pre-test couple relationship. For example, two couples that received positive test results reported frustration as the partners shifted toward caretaking roles even before the people with HD showed any symptoms. In another couple tested, a woman believed to be at risk for HD gained self-esteem from a negative result. With low self-esteem before the test, she had married someone who did not match her ideals in a spouse. After the testing showed she did not have HD, she regretted her decision to marry her husband, clearly leading to relationship deterioration.
Since undesired shifts in roles may contribute to couple relationship breakdown whether the test result is positive or negative, the researchers of this study strongly support post-test counseling. Post-test counseling can help couples find and maintain a new balance that is satisfying to both partners. This counseling should include open communication between the partners, with special attention paid to the desires and worries of each partner.
Conclusions^
It is clear from these studies that the psychosocial impact of predictive testing on the couple relationship is complex, with a number of factors that contribute to both positive and negative outcomes. First, the Richards and Williams study shows that pre-test discussion by the couple can be very helpful to their relationship. Such discussion can better prepare the couple for the test result by encouraging understanding of each other’s expectations of and reactions to the test result. In particular, this pre-test assessment can help identify particular challenges that the couple may face after the testing and may lead to re-consideration of testing in the first place. Complementing the Richards and Williams study, the Decruyenaere study shows the importance of post-test counseling. Post-test counseling can help protect against adverse effects of predictive testing by encouraging open discussion of each partner’s concerns as well as identification of any potential role-shifts that may disrupt the couple relationship.
Further Reading^
- Decruyenaere M, Evers-Kiebooms G, Cloostermans T, Boogaerts A, Demyttenaere K, Dom R, Fryns JP. Predictive testing for Huntington’s disease: relationship with partners after testing. Clinical Genetics. 2004 Jan;65(1):24-31.
This study is not only easy-to-read but also optimistic in its finding that most marital relationships remained the same five years after predictive testing, regardless of the test results.
- Evers-Kiebooms G, Swerts A, Cassiman JJ, Van den Berghe H. The motivation of at-risk individuals and their partners in deciding for or against predictive testing for Huntington’s disease . Clinical Genetics. 1989 Jan;35(1):29-40.
This early study found that the majority of at-risk persons and their partners looked favorably upon predictive testing, although the at-risk individual and his/her partner’s reasons for deciding to take the test varied. This study took place before predictive testing began in 1993; however, the couples’ explanations for deciding on predictive testing are still eye-opening and relevant.
- Huggins et al. Predictive testing for Huntington disease in Canada : Adverse effects and unexpected results in those receiving a decreased risk . 1992 Am J Med Genet 42:508-515.
- Richards F, and Williams K. Impact on couple relationships of predictive testing for Huntington disease: a longitudinal study. American Journal of Medical Genetics Part A. 2004 Apr 15;126(2):161-9.
This is an easy-to-read article that is especially interesting because of its discussion on the benefits of pre- and post-test counseling.
- Soldan et al. Psychological model for presymptomatic test interviews: Lessons learned from Huntington disease . 2000 J Genet Couns 9:15-31.
Studies, in addition to Richards and Williams 2004, that found few negative effects of predictive testing on couple relationships:
- Codori AM, et al. Psychological costs and benefits of predictive testing for Huntington’s disease. 1994 Am J Med Genet 54:174-184.
- Quaid KA, et al. Exploration of the effects of predictive testing for Huntington disease on intimate relationships. 1995 Am J Med Genet 57:46-51.
- Taylor CA, et al. Long-term impact of Huntington disease linkage testing . 1997 Am J Med Genet 70:365-370.
- Tibben A, et al. On attitudes and appreciation 6 months after predictive DNA testing for Huntington disease in the Dutch program . 1993 Am J Med Genet 48:103-111.
-C. A. Chen 5-7-07
More
Click on the link below to hear an audio recording of this article:
The Neurobiology of Huntington’s Disease
Now let’s look at the broader picture and ask a number of questions about the way in which the C-A-G triplet repeat in the genes of people with HD manifests itself in the symptoms of HD. We’ll focus on changes in the brain that take place with the onset of HD.
Podcast: Play in new window | Download
More
basic
Click on the link below to hear an audio recording of this article:
The Inheritance of Huntington’s Disease
Although records of symptoms have been traced as far back as the Middle Ages, it was not until the late 1800s that physician George Huntington first documented the hereditary nature of the disease that bears his name. It was the late onset and hereditary character that distinguished HD from other diseases with similar symptoms. With so many recent breakthroughs in human genome research, we now know quite a bit about the genetic basis of Huntington’s disease. Having a working familiarity with the basic genetics of HD is key to understanding the inheritance and expression of the disease.
What is a gene? Which parts of the genetic code correspond to genes?
The material behind genetic inheritance is a surprisingly simple chemical substance called deoxyribonucleic acid (DNA). Each molecule of DNA is a long, continuous chain of smaller molecules called “bases” that are strung one after the other. The four different bases (abbreviated A, T, C, and G) can be arranged in many different ways. It is the sequential order of these bases that provides the chemical information or “instructions” for inheritance. The DNA ladder can be very, very long, twisted and coiled again around proteins into the shape of chromosomes that sit inside each cell’s nucleus. A chromosome is, in fact, one very long “super coiled” molecule of DNA. (For more on the chemical information in DNA, click here.)

Some regions of DNA contain instructions for making a specific functional product, such as a protein. These regions of functional DNA are called genes. Every long molecule of DNA has some segments that are genes and some that are not. Together, they make up the structures called chromosomes that are found in cell nuclei. (See Figure C-1.) The non-gene segments are sometimes half-jokingly called “junk in the genome” – we still have little idea of what, if anything, these sequences do! Fortunately, though, the genes are always found in the same place on a particular chromosome. The gene responsible for causing HD, for example, is always located on chromosome 4. (Humans have 23 pairs of chromosomes, each of which has been assigned a conventional number. Click here for a look at all 23 pairs.) When scientists say that they have “located” a gene for a disease, they usually mean that they have found a region on one of the chromosomes that codes for a protein that somehow contributes to causing the disease.
What are alleles? How many alleles can there be for a gene and how many copies does each individual have?
Suppose we choose a particular chromosome from two different people and examine the DNA from the same spot on both chromosomes. We will find that the pattern of the bases (A’s, C’s, T’s, and G’s) is similar, but it is often not exactly the same, even if the region is a protein-coding gene. How can a gene code for a product if the pattern is not the same in every person? The answer is that there can be many different versions or variants of a given gene. These different versions of the gene are called alleles. Different alleles of a gene code for the same trait, but they may manifest themselves in different ways. The gene for eye color contains the instructions governing eye pigment, for example, but the specific color is determined by the particular alleles one has. Everyone has the same number of chromosomes and genes, but each person’s genetic code has a unique combination of alleles. This potential for variation explains why we all have similar genomes, yet we still have people of different heights, weights, and faces.
The way in which the Huntington gene varies among individuals is by the number of repeated C-A-G codons it contains. In other words, different alleles of the Huntington gene contain different numbers of CAG codons. It is important to understand that everyone has the Huntington gene, but individuals with Huntington’s disease have a many-CAG version of the gene, one that does not function normally. “Having the HD allele” is somewhat loose terminology, but it is used often and usually implies “having one of the multiple-CAG alleles on the Huntington gene that causes HD.” Within this site, the allele on the Huntington gene with the normal number of CAG repeats (the allele that does not result in HD) is referred to as the non-HD allele. The allele of the Huntington gene with the extra CAG repeats (the allele that does result in HD) is described as the HD allele.
How do genes determine physical traits? What does it mean to say that the HD gene is dominant?
Since we inherit one complete set of DNA from each parent, chromosomes occur in pairs called “homologues.” (Click here for a picture of homologues.) Hence, a gene that is found on a given chromosome actually has a partner on its matching, or homologous, chromosome. This means that a person actually has two copies of every gene, one allele on each of two homologous chromosomes. This feature raises some important questions about how alleles interact and relate to each other.

Alleles can be thought of as having different “strengths.” If two different alleles are present together, the “stronger” one will influence the trait under consideration. This phenomenon is called dominance. A dominant allele influences the resulting trait whether an individual has one or two copies of that allele. In contrast, in order for recessive alleles to be expressed, an individual must always have two copies of the “weaker” allele. (See Table C-1.)
HD is called a dominant trait because individuals with just one copy of the HD allele typically develop HD symptoms. The HD allele (with many CAG repeats) is dominant over the non-HD allele (with few CAG repeats). Again, an individual need have only one copy of the HD allele to inherit the disease. There is also no exact cut-off point for when the number of CAG repeats is considered “abnormal.” (Click here for a table of repeats and their effects.) Occasionally, an individual will have an allele whose CAG codon count falls within a small “gray area” for which the repeat number is slightly higher than normal, but not quite “abnormal.” This version of the allele has a medium strength. Its dominance is said to be “incomplete,” and individuals with this allele may or may not develop the disease.
How are alleles inherited? What are the chances of inheriting the gene?
Since the Huntington gene is not on a sex-determining chromosome, the disease is not sex-linked. In other words, the inheritance and development of Huntington’s disease are not related to an individual’s sex. This means that males and females have an equal chance of inheriting the disease. Males and females with the disease are also equally likely to pass it on to their children.
Every person inherits two copies of the Huntington gene, one from each parent. Likewise, every person will also pass one of these two copies to each child. The chances of giving either of these two alleles to a child are equal (50/50). A person with Huntington’s disease has one non-HD allele and one HD allele. Hence, there is a 50% chance that the non-HD allele will be passed on and a 50% chance that the HD allele will be passed on. This means that each child of an individual with HD has a 50% chance of getting the HD allele. Individuals with a chance of inheriting the disease are sometimes described as “at-risk.” At-risk individuals have the option of undergoing genetic testing, which shows whether their “50% risk” of developing HD is in reality nearly 0% or nearly 100%. (For more information about genetic testing for HD, click here.)
Individuals without any copies of the HD allele do not have HD, and these individuals are very unlikely to pass HD on to their children. They have two non-HD alleles, and the child will always receive one of these two alleles. The only exception is in the case of a new mutation, a heritable change in a person’s DNA. Very rarely a mutation will occur so that a child’s allele differs from that of the parent from whom it was inherited. Only very rarely, therefore, does an individual without an HD allele have a child with HD. For the same reason, Huntington’s disease does not typically “skip” generations. That is, we do not observe families in which a grandparent and grandchild have HD but the child’s parents do not. If such a pattern were observed, it would be most likely that one of the parents has an HD allele, but has not yet developed symptoms of the disease.
Are my children at risk?
Let’s switch gears and think about this question from the perspective of the child of a person with HD. The child inherits one allele from each parent. The parent without HD has two non-HD alleles, so the allele from this parent will be non-HD regardless of which one is inherited. The parent with HD has one non-HD allele and HD allele. There is an equal probability of passing either of these alleles to the child. Thus the child has a 50% chance of getting the non-HD allele and a 50% chance of getting the HD allele. Since the chance of getting an HD allele is one in two, the child has a 50% chance overall of inheriting the disease. (See Figure C-2.)
What if you discover that you are the grandchild of a person with HD, and your parent who is at risk chooses not to be tested? Recall that you have two copies of the Huntington gene. One copy (allele) will come from the parent who is not at risk. This copy will always be non-HD and does not affect your chances of getting the disease. The second copy comes from your at-risk parent. Since this parent is the child of an individual with HD, he or she has an equal chance of having either two non-HD alleles or one non-HD and one HD allele. (We found this out in Figure C-2.) In the first case, this parent has two non-HD alleles and you will not inherit the disease regardless of which of the two non-HD alleles you get. In the second case, the parent has one HD allele and one non-HD allele. Here, you will inherit the disease if you get the HD allele, but not if you get the non-HD allele. Out of the four possible outcomes, exactly one results in your having a copy of the HD allele. This represents a 25% chance that you have inherited the disease. (See Figure C-3.)
There has never been any history of HD in my family…how could the HD allele just suddenly appear?
Remember that HD alleles are distinguished by the number of CAG codons they contain. The number of repeats is not fixed between generations, and it is possible that the number of repeats changes when cells divide. Usually this change results in a larger number of repeats, although occasionally the number of repeats decreases. No one is sure exactly what causes the number of repeats to multiply, but there is some evidence that codon numbers expand as a result of DNA copying inaccuracies during sperm formation. When DNA is copied, it is reproduced in small sections that are strung together later to make the long, continuous strands of DNA in chromosomes. There is some speculation that codon repeats could expand if these pieces are not hooked together correctly. (For more information about mutations, click here.)
A person who has a “normal allele” with a borderline number of repeats (typically between 36 and 39 copies of CAG) may produce a sperm or egg cell that contains an allele with a few additional codons. On rare occasions, these extra codons may be just enough to cause the child inheriting the allele to have an abnormal repeat number. One researcher speculated that about 10% of HD cases are caused by such changes in repeat number. Although in most cases the HD allele is not inherited in this way, this possibility explains how HD sometimes seems to just “appear” in a family.
If I have the gene, what does that mean? Will it definitely express itself, and if so, when will this happen?
Historically, an individual known to have an HD allele almost always developed symptoms of the disease, unless he or she died of some other cause prior to onset of symptoms. Individual cases have varied greatly in severity and in rate of progression for reasons that are still not yet fully understood. As a very general rule, the typical age of onset for adult-onset HD is between the ages of 30 and 50. In most instances, individuals live with the disease for 10 to 25 years. Several studies indicate that the number of CAG codons plays a role in how soon symptoms appear. The general trend appears to be “the greater the number of repeats, the earlier the onset of the disease” although there is considerable variation. There is also evidence to suggest that the average age of onset is later for individuals who inherited the HD allele from their mothers than for individuals who inherited the allele from their fathers. It follows then that, as a general rule, onset occurs earlier when the HD allele is inherited via one’s father.
About 10 percent of all HD cases are classified as the juvenile form, which has an age of onset between infancy and 20 years. The juvenile form generally occurs when the number of CAG codons is especially large (on the order of 55 and above). Individuals with juvenile HD have different symptoms, such as rigidity, seizures, and dementia, than do individuals with adult HD. In addition, the progression of juvenile HD is usually much more rapid. (Click here for more about juvenile Huntington’s disease.)
Figure C-4 shows the correlation between increasing number of CAG repeats, from 39 to 50, and decreasing age of onset. The shaded bars show the median age of onset for individuals with a given number of CAG repeats. The exact numbers used for this graph are shown in Table C-2. Table C-2 also shows, in its third column, a way to represent the range in age of onset that occurs at each number of repeats. The figures in the third column correspond to 85% confidence intervals (C.I.) around the average age of onset. A confidence interval means that for a given number of repeats, we can be 85% sure that the actual age of onset lies within the given age range. This range is represented below in Figure C-4 by yellow bars. Please note that these range figures must be interpreted carefully: they specifically do not imply that an individual who remains symptom-free throughout a given range is exempt from HD. Any particular individual may very well develop symptoms at either an earlier or later age than those shown in the table.


For further reading
- “Huntington’s Disease: A Brief Explanation” International Huntington Association. Online.
- “Huntington’s Disease” Online Mendelian Inheritance in Man. Online.
- “GuideLines for Genetic Testing for Huntington’s Disease.” Huntington’s Disease Society of America. Online.
-A. Hsu, updated 7-1-04
Podcast: Play in new window | Download
More
Proteins and what they do
Proteins are very important molecules to all forms of life. They are one of four of life’s basic building blocks; the other three are carbohydrates (sugars), lipids (fats), and nucleic acids (DNA and RNA).
More
Scenes 1-5
16th Century
Paracelsus, a Renaissance alchemist (1493-1541), coins the term “chorea” to describe the dance-like, uncoordinated movements that are now known to be symptomatic of HD
1686
English physician Thomas Sydenham attempts to classify different types of chorea and describe their causes.
1630s
English colonists in Massachusetts, Connecticut, and New York (especially Long Island) use names such as “that disorder” and “Saint Vitus´ dance” to describe HD.
1692
The Salem Witch Trials occur in Salem, Massachusetts. Some of the “witches” are now believed to have had HD. Their choreic movements and odd behavior were seen as possession by the devil.
1840s
For the first time, HD is described in the medical literature as “chronic hereditary chorea.” Physicians in the United States, England, and Norway write about people with involuntary movements and mental disturbances that were inherited from a similarly affected parent. Three separate accounts are recorded, all by young physicians.
Scenes 6-10
1872
George Huntington writes a landmark paper entitled “On Chorea.” Using personal accounts of his father´s patients, Huntington provided a classic description of HD´s symptoms and emphasizes HD´s hereditary nature. Significant interest in HD, especially its genetic component, occurs due to George Huntington´s paper, “On Chorea” (1872).
1910-11
The American eugenicist Charles B. Davenport writes Heredity in Relation to Eugenics (1911), in which he uses genetic diseases, including HD, to argue in favor of compulsory sterilization and immigration restriction for those afflicted with HD. Davenport founds the Cold Spring Harbor Biological Laboratory and Eugenics Record Office in 1910 to track families with inherited disorders, and he produces what is, at the time, the largest study of families with HD.
1910-11
Researchers first note the deterioration the central region of the brains of patients as the disease progresses. They identify the caudate nucleus as the central target of brain cell death.
1950s
An upsurge in publications on HD research occurs with the growing interest in human genetics and the 1953 discovery of DNA´s structure by Watson and Crick.
1955
Americo Negrette publishes a book describing communities in Lake Maracaibo, Venezuela, with unusually high numbers of individuals affected by HD.
Scenes 11-15
Late 1950s
Arvid Carlsson and Oleh Hornykiewicz, two European scientists, make the breakthrough discovery that dopamine pathways between neurons are destroyed in Parkinson´s disease patients. Since the symptoms of Parkinson´s disease are nearly the opposite of those of HD, the scientists hypothesize that decreasing HD patients´ dopamine levels might be a key step in treating the disease.
1966
The first Department of Neurobiology is established at Harvard University. Ntinos Myrianthopoulous writes a review article decrying the lack of knowledge of HD.
1967
Famous poet and songwriter Woody Guthrie dies of HD. Guthrie´s wife, Marjorie, creates the Committee to Combat Huntington´s Disease (CCHD), now called the Huntington´s Disease Society of America (HDSA), to provide public health outreach on HD.
1970
The Society for Neuroscience (SFN) is a nonprofit organization dedicated to study of the brain and nervous system. SFN, founded in 1970, has grown from 500 members to more than 36,000. Society of Neuroscience is the world’s largest organization of scientists devoted to the study of the brain.
1972
The International Centennial Symposium on Huntington´s Disease is held on the hundredth anniversary of George Huntington´s historic publication (See 1872). The Symposium aims to gather all HD researchers and assess the current state of knowledge, generating new optimism for HD research.
Scenes 16-20
1972
Thomas L. Perry finds diminished levels of GABA in the brains of HD patients.
1973
John Meeks and Natalie Stein suggest that the HD allele causes premature aging.
1974
Milton Wexler establishes the Foundation for Research in Hereditary Disease, which will later become the Hereditary Disease Foundation.
1976
Joseph T. Coyle develops the first rat model of HD by using kainic acid. The rats exhibited HD-like symptoms such as decreased weight, motor dysfunction, brain atrophy, neuronal inclusions and other cognitive impairments.
1977
The Congressional Commission for the Control of Huntington´s Disease and Its Consequences is held to develop a comprehensive report on HD in the United States.
Scenes 21-25
1978
The Second International Centennial Symposium on Huntington´s Disease is held to review progress since the 1972 Symposium. The sheer volume of research that is accomplished over the six years indicates a heightened interest in HD.
Late 1970s
Researchers find evidence that HD affects cells all over the body, not just in the brain.
1979
Mike Connely establishes the National HD Research Roster at the Indiana University School of Medicine.
Nancy and Tom Chase go to Venezuela for an exploratory visit to the Lake Maricaibo area, a hot spot for HD (see 1955).
1980
The National Institute of Neurological Disorders and Stroke (NINDS) funds the first two “Centers Without Walls” in Boston (Harvard/Mass General Hospital) and Baltimore (Johns Hopkins University).
1981
Nancy Wexler begins her fieldwork in the Venezuelan communities around Lake Maracaibo, a hot spot for HD.
Scenes 26-29
1983
Scientists discover a gene marker linked to HD on the short arm of chromosome 4, which indicates that the Huntington gene is also located on chromosome 4. Predictive linkage testing is introduced to assess the likelihood of contracting HD.
1993
The location of the Huntington gene is discovered at the 4p16.3 gene site on chromosome 4. The gene is found to contain codon C-A-G in varying numbers. An abnormal number of CAG repeats turns out to be a highly reliable way to tell whether someone has the allele for HD.
2000
The Huntington´s Disease Advocacy Center (HDAC) is created to provide information and support for people with HD and their families.
2001
HOPES is a team of faculty and undergraduate students at Stanford University dedicated to making scientific information about Huntington´s disease (HD) more readily accessible to the public. Our goal is to survey the rapidly growing scientific literature on HD and to consolidate this information into a coherent, reliable web resource that reflects current scientific understanding of HD.
Scenes 30-35: Notable People and Places
Philippus Aureolus Theophrastus Paracelsus Bombastus von Honenheim
1493-1541
Paracelsus was a notable alchemist and reformer during the Renaissance period. He introduced the name chorea sancti viti (Latin for “St. Vitus´ dance”) to describe a peculiar disease characterized by writhing, sporadic movements. Most likely due to the mass hysteria and religious superstition of the time, this “dancing mania” had reached epidemic proportions in Europe. It is now thought that many of the sufferers may have experienced epileptic seizures or ergot poisoning. Near the end of Paracelsus´s lifetime, the spread of the disease began to slow, the symptoms became milder, and Paracelsus termed this new form “chorea naturalis,” or chorea due to natural causes.
Thomas Sydenham
1624-1689
Thomas Sydenham was an English physician who is considered one of the most important revivers of Hippocrates´ views. He stressed careful observation and bedside attendance, and he remarked keenly on many symptoms commonly associated with HD. He noted, for instance, “The hand cannot be steady for an instant. It passes from one position to another, however the patient may strive to the contrary.” He believed that these movements were caused by “some humor falling on the nerves, and such irritation causes the spasm.” Today, however, Sydenham chorea refers to chorea that is associated with rheumatic fever, even though Thomas Sydenham was an English physician who is considered one of the most important revivers of Hippocrates´ views. He stressed careful observation and bedside attendance, and he remarked keenly on many symptoms commonly associated with HD. He noted, for instance, “The hand cannot be steady for an instant. It passes from one position to another, however the patient may strive to the contrary.” He believed that these movements were caused by “some humor falling on the nerves, and such irritation causes the spasm.” Today, however, Sydenham chorea refers to chorea that is associated with rheumatic fever, even though Sydenham never explicitly made that link.
George Huntington
1850-1916
George Huntington, an American physician, was only twenty-two years old when he submitted his famous paper “On Chorea” (1872) to The Medical and Surgical Reporter. Much of the paper drew from the written observations of his father and grandfather, both physicians who had noticed the involuntary shaking of some patients. The paper gained Huntington instant notability because, in the words of Sir William Osler, “In the history of medicine there are few instances in which a disease has been more accurately more graphically, or more briefly described.” Huntington was able to explicitly point to genetic inheritance as the mode of transmission, and he noticed that the first symptoms usually appear at an adult age and that they are usually accompanied by mental decline as well. It is due to these significant observations and conclusions that “Huntington´s disease” bears George Huntington´s name.
Woody Guthrie
1912-1967
Woody Guthrie was one of the most famous Americans with HD. Born in Okemah, Oklahoma, Guthrie gained fame in the 1930s and 1940s as a folk singer and radio entertainer. He was known for putting political and social commentary in the lyrics of his music, and he often celebrated the plight of the American laborer. In his songs, Guthrie includes references to many of the 20th century´s most historic events, including the Great Depression, the “Dust Bowl” migration, World War II, and the Cold War. His most famous songs include “This Land Is Your Land,” “Grand Coulee Dam,” and “I Ain´t Got No Home.”
Guthrie´s mental state began to deteriorate in the early 1950s. His memory declined, and his behavior became unpredictable. He left his wife, Marjorie, and his home in New York to marry a woman twenty years his junior in California. However, due to his mental state, Guthrie was eventually forced to return to New York, where he was placed in one hospital after another. HD
The Wexler Family
The Wexler family is inextricably tied to the history of Huntington´s disease research. In 1968, Leonore Wexler was diagnosed with HD, which inspired her two daughters, Nancy and Alice, and her husband, Milton, to become involved in the search for a cure for HD.
Milton Wexler, a prominent psychologist, is responsible for bringing world renown researchers together to focus on HD research. He founded the Hereditary Disease Foundation, which funds HD research and sponsors workshops for scientists to share ideas.
Nancy Wexler has played a pivotal role in the scientific research of HD. She pioneered the fieldwork in Lake Maracaibo, Venezuela that led to the discovery of the Huntington gene (see Lake Maracaibo, Venezuela) and has since helped other researchers map genes responsible for Alzheimer´s disease, kidney cancer, manic depression, and other disorders. She served as the Hereditary Disease Foundation´s president, and is currently a Professor of Neuropsychology at Columbia University.
Alice Wexler, a teacher, writer, and historian, chronicled her family´s journey in the insightful book Mapping Fate: A Memoir of Family, Risk, and Genetic Research.
Huntington’s disease in Lake Maracaibo, Venezuela
In the early 1950s, Dr. Amerigo Negrette first diagnosed Huntington´s disease in Lake Maracaibo, Venezuela. Working as a rural physician, Negrette was perplexed by the fact that many townspeople often appeared drunk, staggering and weaving at all hours of the day. He learned from locals that these people were not drunk, but instead suffered from a disease referred to as el mal de San Vito, or the sickness of Saint Vitus. After visiting many people with the sickness, Negrette diagnosed the disease as HD. He discovered that HD ran deep in the community; people with the illness were interrelated and had common ancestry. In 1963, he published a book entitled Corea de Huntington: Estudio de una Sola Familia a Traves de Varias Genereaciones describing HD in his community. The world learned of this tragic occurrence when Negrette´s work was presented at the 1972 Centennial Symposium.
In 1981, Dr. Nancy Wexler led a team of scientists to study HD in Lake Maracaibo. Their original goal was to find an HD homozygote (an individual who has inherited two copies of the HD allele), but the team also ended up collecting blood samples from as many HD sufferers as they could find and test. These samples played a key role in the discovery of a genetic marker for HD in 1983 and led to the creation of a community pedigree, the largest of its kind in the world.
More
Click on the link below to hear an audio recording of this article:
Juvenile Huntington’s Disease
Juvenile HD is a form of Huntington’s disease that affects children and teenagers. Like the adult form of the disease, juvenile HD is hereditary in nature. Because of its hereditary character and early age of onset, a child with juvenile HD may also have a parent or other close family member who is affected by adult-onset HD at the same time. This tendency to affect multiple generations simultaneously places an even greater strain upon families who are affected by juvenile HD.
What is juvenile HD?

Although juvenile HD and adult-onset HD both result from an altered form of the same gene (the Huntington gene), the symptoms of juvenile HD are very different from those of adult-onset HD. Individuals with juvenile HD often become stiff or rigid in their movements (instead of having chorea), and about one third of them have recurrent seizures. As with adult-onset HD, individual cases of juvenile HD vary greatly, and different children often have different symptoms. As a result, cases of HD are classified as the juvenile or adult form based upon age of onset, and not by symptom. Any case of HD where the onset occurs before the age of 20 is considered to be of the juvenile form, regardless of the symptoms present.
Although the number of CAG codon repeats in a particular segment of the Huntington gene does not accurately predict the age of onset, generally more repeats correspond to an earlier age of onset. This tendency is especially true in cases of juvenile HD, where most individuals have between 80 and 100 CAG repeats. The earlier the onset of juvenile HD, the faster it usually progresses. In general, progression of the disease is more rapid than in adult-onset HD. Often, death from juvenile HD occurs within 10 years of onset, as opposed to 10-25 years in adult-onset HD.
What causes the large CAG repeat numbers seen in juvenile HD cases?
Children with juvenile HD usually have a larger number of CAG repeats in a particular segment of the Huntington gene than do individuals with adult-onset HD. In many cases, these children also have many more CAG repeats compared to the parents from whom they inherited the HD allele. The exact cause of repeat expansion is still unclear. At one point, it was thought that the DNA from the unaffected parent might somehow contribute to the development of juvenile HD. A non-HD allele from the unaffected parent could potentially “aggravate” the Huntington gene and somehow cause the large increase in repeat numbers characteristic of juvenile HD. Given that juvenile HD is so rare, if there existed an allele that aggravated the disease, it would necessarily be rare as well. A case study in the 1960’s showed a man with adult-onset HD who had affected children with two different women. In order for this to occur, both of the mothers must have had the rare “aggravating allele,” a highly improbable occurrence. This finding made it seem unlikely that DNA from the unaffected parent was contributing to the expanded repeats.

(For more information on DNA, click here.)
Along with this process comes the possibility that a mistake is made somewhere during the copying procedure. Such “mistakes” are very common during DNA replication. One such mistake might cause the number of codon repeats to increase. Since the formation of sperm involves millions of cell divisions more than the formation of eggs, the number of opportunities for triplet expansion during DNA replication is much larger in males than in females. Hence, it is possible that adult males are more likely to pass alleles with expanded repeat numbers to their children. We will call this the “paternal triplet expansion hypothesis.” This hypothesis could explain why in most cases (about 70-90% of them), individuals with juvenile HD have inherited the HD allele from their fathers rather than their mothers.
How are large repeat numbers related to the increased severity of juvenile HD?
Individuals with early-onset HD usually have a number of CAG repeats in a particular segment of the Huntington gene that is much larger than the number of repeats seen in adult-onset HD. The largest number of CAG repeats seen thus far is around 250, but most individuals with juvenile HD have between 80 and 100 repeats.

(For another example of correlation and causation click here.)
Most individuals with juvenile HD experience an age of onset that is much younger than that of their affected parents. They also often face a much more rapid progression of the disease. This occurrence is described as genetic anticipation, where a disease increases in severity in successive generations, and a parent can produce a child with a more severe form of a disease. In the case of HD, the expanded section of triplet repeats provides a possible (though still unconfirmed) explanation for the pattern of anticipation seen in HD inheritance. As the number of repeats grows between generations, the severity of the disease increases, and individuals experience an earlier age of onset and a more rapid development of the disease.
How is juvenile HD inherited?
Juvenile HD is caused by the same gene that causes adult-onset HD. The version of the Huntington allele causing early-onset HD usually has, however, a greater number of CAG repeats. Because the early-onset and late-onset forms depend upon the same gene, early-onset HD is inherited in the same manner as adult-onset HD. (To read about how the HD allele is inherited, click here.)

Due to the rapid progression of the disease, most individuals with juvenile HD do not survive to bear children of their own. For those who do, however, their children have the same 50% risk of inheriting the HD allele as the children of individuals with adult-onset HD. The number of CAG repeats in the Huntington gene of an individual with juvenile HD is normally very high (even compared to that of individuals with adult-onset HD). Since repeat numbers tend to increase rather than decrease in successive generations, it is likely that the child of such an individual will have a similar or larger number of repeats if he or she inherits the altered allele. Given the correlation between repeat number and age of onset (discussed in the previous section), it is very likely that the child will also develop juvenile HD. In short, the child of an individual with juvenile HD has a 50% chance of inheriting the HD allele. If the child does inherit the altered allele, he or she is very likely to develop juvenile HD.
What are the early signs of juvenile HD?
Although by definition juvenile HD begins at an early age, most children are able to walk and talk at a normal age before symptoms start to appear. The signs of juvenile HD are often subtle and difficult to distinguish from the normal “growing pains” that children experience. A major sign of onset is a continuing decline in school performance. Other indications include subtle changes in handwriting, difficulty learning new things, and small problems with movement. Some common movement problems include slowness, clumsiness, rigidity, tremor, and muscular twitching, or myoclonus. Parents often notice that their children fall more often and are less coordinated than they used to be.
Every case of juvenile HD is unique, and it is possible that individuals experience different symptoms depending on the age of onset and exact number of CAG repeats. However, many parents of children with HD have said that the most noticeable aspect of onset is change. Parents might notice personality changes, new problems with coordination, behavioral changes, new speech difficulties, and changes of pace in learning. For example, a child who was once very good at sports has become clumsy in recent months, or a previously well-behaved student is suddenly causing trouble at school. A mother of two children with HD described her perception of the changes within her family members:
“Following the diagnosis of HD in the first child, I began, of course, to observe the other family members very closely – ever vigilant for signs of HD. Some things, such as moodiness, speech problems, or hyperactivity could have been interpreted as early symptoms of HD. It became apparent that the clue seemed not to be the action itself, but rather, whether or not those things had always been present or if they represented a definite change.”
What symptoms are common to both juvenile HD and adult-onset HD?
Both the early- and adult-onset forms of HD are characterized by what is called dementia, a progressive loss of mental function. Many individuals also seem to undergo personality changes. Some changes, such as increased irritability and bad temper outbursts, are sometimes due to the difficulties of dealing with the disease rather than actual clinical symptoms. Often, people with HD experience frustration when realizing that they can no longer do things they once could. Sometimes, however, these personality changes are a more direct result of the disease. Such symptoms may be alleviated with medication.

For an explanation of these differences click here.
How are the symptoms of juvenile HD different from those of adult-onset HD?
The most notable symptomatic distinction between the two forms of HD is that many individuals with juvenile HD do not experience the chorea that is so commonly associated with the adult-onset form. Instead of exhibiting the dance-like movements of chorea, affected children are often rigid and stiff. Generally, children with a younger age of onset are less likely to experience chorea. Chorea is more likely to be present in individuals who have an age of onset from 15-18 years. It seems that individuals with juvenile HD who have a later age of onset are more likely to experience symptoms that resemble those of adult-onset HD.
About 25-30% of individuals with early-onset HD also experience recurring seizures, a symptom that is uncommon in the adult-onset form. Seizures experienced by children with HD are usually generalized, meaning that they are caused by electrical discharges that affect both sides of the brain and often involve a loss of consciousness. However, some children also develop partial seizures, which involve discharges in just one part of the brain and may or may not involve a loss of consciousness.
The generalized seizures experienced by HD children are usually what are called tonic-clonic seizures. Generalized tonic-clonic seizures (or grand mal seizures) consist of both tonic and clonic phases. During the tonic phase the body is rigid, and often the child falls to the ground. The clonic phase follows the tonic phase and is usually associated with convulsive movements or rhythmic jerking motions. The child typically loses consciousness for a variable period of time.
Some children also develop myoclonic seizures, which involve sudden, brief jerking movements, or myoclonus. These seizures vary greatly in their severity and frequency. Myoclonus should not be mistaken for seizures – the term myoclonus refers to the jerking symptom itself, which can have many causes. It is only when myoclonus is caused by abnormal brain activity that it is properly called myoclonic seizure. Many children with HD experience myoclonus that is not related to seizures.
What parts of the brain are affected in juvenile HD?
At autopsy, individuals who have died from juvenile HD show an even more widespread pattern of brain degeneration than that seen in adult-onset HD. As in the adult form, there is severe degeneration of the caudate and putamen. (See Figure D-4.) The caudate and the putamen are responsible for regulating voluntary movement, and it is thought that damage to these parts of the brain is responsible for many of the movement problems — especially the chorea — that individuals with HD experience. (See Figure E-1.)
A characteristic that is seen more often in the juvenile form than in the adult form is extreme gliosis of the globus pallidus (Figure E-1). Gliosis is excess growth of what are called spider cells (see Figure E-2) — cells that normally provide supporting and protective tissue for nerve cells. Some individuals with adult-onset HD experience rigidity (instead of chorea), and case studies of several of these individuals have also shown damage to the globus pallidus. Hence, it is thought that abnormality of the globus pallidus may be responsible for the rigidity seen in juvenile HD.
Analysis of juvenile HD brains shows damage to many areas, but the pattern of damage is not consistent between individuals. Loss of neurons in the Purkinje cells and granule cells of the cerebellum is often seen in the juvenile but not the adult form. Other areas of damage sometimes include the dentate nucleus, hippocampus, and neocortex. The dentate nucleus is responsible for rapid movements, and the hippocampus deals with the transfer of information from short-term to long-term memory. The neocortex constitutes about 85% of the brain’s total mass, and it is believed to be responsible for higher cognitive functions, such as language and memories. It is currently not known how damage to these areas of the brain manifests itself as symptoms in people with juvenile HD.
What treatments are recommended specifically for juvenile HD?
Anticonvulsant drugs are usually prescribed to help prevent and control the seizures that occur in children with juvenile HD. Finding the right combination and amount of drugs is not an easy process, and often the optimal treatment varies over time and between individuals. In many cases, caregivers know the most about the child’s reactions to specific drugs, making it very important for the doctor and caregivers to communicate frequently about which drugs and doses are working and which are not.

Myoclonus and jerking motions are usually not treated unless they are very severe (for example, if they cause the child to fall frequently or reduce the child’s ability to take in food). Antimyoclonic drugs such as valproate are sometimes prescribed to treat myoclonic jerks.
Side effects of seizure drugs can include drooling, sleepiness, and a general sense of confusion. However, the most significant concern of seizure medications is their potential to aggravate other juvenile HD symptoms. Some drugs may cause increased swallowing problems, drowsiness, and coordination difficulties. Many children with HD also have a poor tolerance of anticonvulsant drugs. Generally, physicians attempt to minimize the seizures as much as they can without lowering the quality of life in other areas. Achieving this ideal balance often requires trying many different drugs and prescribing less than the maximum dosage of each particular drug. Although the children may still have occasional seizures, many parents consider this treatment more acceptable than the alternative: prescribing a higher dosage to eliminate seizures but worsening the child’s other symptoms.
Physical therapy is recommended to ease rigidity and to prevent degeneration (atrophy) of unused muscle. For some individuals, pool therapy especially helps to loosen tight muscles. Pool therapy involves exercises that are done while the individual is submerged in warm water. The warm temperature is soothing for muscles, and the buoyancy of the water makes motion require less effort, enabling patients to strengthen muscles gradually.
Drugs are sometimes prescribed to control other symptoms, such as rigidity and difficulty sleeping. Counseling and medication sometimes help with behavioral and psychological symptoms. Many times individuals with juvenile HD respond poorly to drugs that are commonly prescribed for adult-onset HD. Hence, with each new drug or dosage, the child should be monitored carefully for side effects, such as increased drowsiness or poorer performance in school. The most effective combination of treatments is different for every individual with juvenile HD, and this optimal care can be achieved only when the doctor and caregivers work together to discover what is best for the child.
Table E-1 gives an abbreviated summary of juvenile HD.
For further reading
- “Juvenile Huntington Disease,” Huntington Society of Canada. Online.
An excellent resource for juvenile HD; one of the few souces that focuses only on juvenile HD.
- Byers, R. K., and J. A. Dodge (1967) “Huntington’s chorea in children. Report of four cases.” Neurology 17: 587-96.
A paper describing the symptoms, treatment, and diagnoses of four children with juvenile HD.
- Byers, R. K., F. H. Gilles, and C. Fung (1973) “Huntington’s disease in children. Neuropathologic study of four cases.” Neurology 23: 561-9.
A very detailed paper that analyzes the brains of four individuals who died from juvenile HD and compares them to similar neuropathologic studies.
- “Case 117 — Progressive Movement Disorder,” University of Pittsburgh School of Medicine, Department of Pathology. Online.
A case study of the diagnosis of a girl with juvenile HD.
A. Hsu, 2-25-02
Podcast: Play in new window | Download
More
base mass refined range nucleus basic control CNS ganglia
Welcome
Introduction
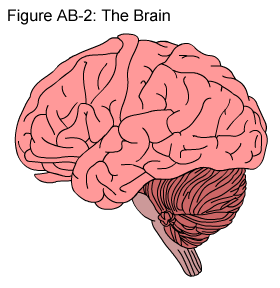
The brain is a complex organ with many components. These multiple components work together to maintain basic life processes, like breathing, body temperature and blood pressure, as well as higher functions like creative thought and emotions. This is an introduction to some of the basic terms used when describing the brain and its major parts.
Central Nervous System

The brain is a part of the central nervous system (CNS). It receives information from other parts of the body via the spinal cord and the peripheral nervous system and uses this information to control the body.
Brain Cells
The brain is made up of two types of cells: neurons and glial cells. Neurons are nerve cells. They typically consist of dendrites that receive information, a cell body, and an axon that is used to transmit information throughout the nervous system. Glial cells have multiple functions, which include structurally supporting neurons, repairing the CNS, and regulating the biochemical balance of the brain. The blood-brain barrier is composed of astrocytes, a specific type of glial cell. This barrier prevents many substances in the blood from entering the brain.
Directions
The top of the brain is called the superior side, and the bottom is called the inferior side. Structures near the center are referred to as medial, and farther away from the center are referred to as lateral. The term anterior means ‘in front,’ while posterior means ‘behind.’
Protection

The brain is surrounded and protected by the rigid, bony skull and three membranes, or meninges. The tough, fibrous outer membrane is the dura mater. The intermediate membrane, named the arachnoid, is thin and weblike. The pia mater is the innermost covering and is the most delicate. It is molded to the shape of the brain. The cerebrospinal fluid (CSF) surrounds the brain and spinal cord and flows through open chambers in the brain, known as ventricles, and out an opening to the spinal cord. The brain actually floats in the shock-absorbing CSF, and is thus protected from trauma. The CSF also brings nutrients to the brain and removes wastes.
Cerebral Cortex
The outermost and top layer of the brain is the cerebral cortex. The cerebral cortex is the most recently evolved and most complex part of the brain. As one moves lower into the brain, the parts have increasingly primitive and basic functions and are less likely to require conscious control.
Brain Hemispheres
The cerebral cortex is divided down the middle, from front to back, into hemispheres, or halves. Each hemisphere has different functions. The left side of the cerebral cortex controls the right side of the body and speech. The right side controls the left side of the body and the perception of spatial relationships, such as where one’s foot might be located in relation to the ground.
Brain Tissue

White matter, which consists of neuronal axons, makes up the inner core of the hemispheres. The outer layer is made up of gray matter, which consists of neuron cell bodies. The cortex contains ridges (gyri) and valleys (sulci).

The hemispheres are separated by a deep groove, but are connected deep in the brain by the corpus callosum, a thick bundle of nerve fibers through which information is passed between the left and right hemispheres of the brain.
Lobes of the Brain
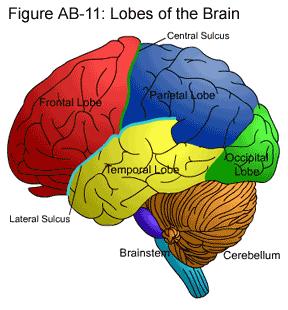
Two sulci – the central sulcus and the lateral sulcus – are used to divide each hemisphere into four sections known as lobes: the frontal lobe, parietal lobe, temporal lobe, and occipital lobe. (Below follows more information on each lobe.)
All of the lobes also contain areas for which specialized functions have not yet been identified. These areas are known as the association cortex and are thought to be involved in complex, higher-level mental activity.
Frontal Lobe

The frontal lobe has a major role in the planning and execution of movements. It contains the pre-frontal, pre-motor and motor areas, listed from front to back.
The pre-frontal cortex is particularly associated with higher level thought, decision-making and planning. It has a significant inhibitory role over impulses and actions. People with HD sometimes have degeneration of the prefrontal cortex, leading to increased impulsiveness and changes in behavior.
The pre-motor and motor cortices process and transmit information regarding body movement. There are two pathways connecting the motor cortex and the basal ganglia to coordinate movement. As nerve cells in the basal ganglia die as a result of HD, both of these pathways are eventually damaged.
Parietal Lobe

The parietal lobe is separated from the frontal cortex by the central sulcus. It lies posterior to (behind) the frontal lobe and superior to (above) the temporal lobe. The parietal lobe contains the primary sensory cortex through which sensations, such as touch and pressure, are felt. In addition, it has a key role in spatial orientation and information processing.
Temporal Lobe

The temporal lobe is located inferior to (below) the frontal and parietal lobes. It is primarily involved with auditory processing and memory.
Occipital Lobe

The occipital lobe is located posterior to (behind) the temporal lobe and is the visual center of the brain. Visual information from the eyes is processed here.
Limbic System

The limbic system wraps around the brain stem and is beneath the cerebral cortex. It is a major center for emotion formation and processing, for learning, and for memory. The limbic system contains many parts, including the cingulate gyrus, a band of cortex that runs from the front of the brain to the back, the parahippocampal gyrus, the dentate gyrus, and most notably, the hippocampus and amygdala. The hippocampus is involved in memory storage and formation. It is also involved in complex cognitive processing. The amygdala is associated with forming complex emotional responses, particularly involving aggression. The limbic structures are also connected with other major structures such as the cortex, hypothalamus, thalamus, and basal ganglia.

HD affects the communication of the limbic system with the frontal lobes by damaging the caudate nucleus, a relay station between them. As the connections degenerate, the activity-initiating frontal lobes are disconnected from the emotion processing center of the brain, producing apathy, a common symptom of HD.
Basal Ganglia
Deep in the gray matter of the brain are the basal ganglia. The basal ganglia, along with the cerebral cortex and diencephalon, compose the region of the brain called the forebrain. The basal ganglia connect to the cortex and thalamus and organize muscle-driven “motor” movements of the body. They are the parts of the brain most affected by HD and many of the symptoms of HD result from damage to them.
The major divisions of the basal ganglia are the caudate nucleus, putamen, globus pallidus and substantia nigra.
Caudate Nucleus
The caudate nucleus is a collection of neuronal bodies that connects to many parts of the brain. Together with the putamen, it comprises the neostriatum. Its neurons are most affected by HD, and the deterioration of its connections result in behavioral changes and the inability to control emotions, impulses, thoughts or movements. Damage to the caudate may also result in the inability to experience intense feelings of embarrassment, guilt or shame. Individuals with caudate damage may become “stuck” on one idea or activity, resulting in a lack of self-awareness and inability evaluate their own behavior. This decreased self-awareness may cause individuals to be unaware of mistakes that are evident to others. It may also impair their ability to experience a range of subtle emotions and see other points of view, making social and personal relationships more difficult.
The caudate organizes and filters information that is sent to the frontal lobe, particularly information from the limbic system. Caudate malfunction can affect the functioning of the frontal lobes through a lack of information or an improper amount of information. The caudate assists the frontal lobes in prioritizing the transfer of information to other parts of the brain. Damage to the caudate makes it difficult for people with HD to prioritize tasks and organize their day, as well as to handle many simultaneous stimuli. Along with the putamen, the caudate also controls voluntary movement.
Putamen
The putamen and caudate are collectively known as the neostriatum. Together, they control voluntary movement. The cells of the neostriatum are the first to die as a result of HD, disrupting both the indirect and direct pathways controlling movement. The death of neostriatal neurons in the direct motor pathway leads to the under-stimulation of the motor cortex, causing the slow speed of motor movement often seen in persons with HD. The death of neostriatal neurons in the indirect motor pathway leads to over-stimulation and chorea.
(For more information about these motor pathways, click here.)

Neostriatum / Striatum
The neostriatum, comprised of the caudate and putamen, transmits information to the subthalamic nuclei that modulate motor control. Together, the neostriatum and globus pallidus make up the striatum.
Globus Pallidus
The globus pallidus relays information from the caudate and putamen to the thalamus and is part of the striatum. In HD, both its external and internal regions suffer neuronal loss. Gliosis, the excessive growth of cells that normally support and protect neurons in the globus pallidus, is implicated in juvenile HD. Individuals who experience rigidity rather than chorea may also have damage to the globus pallidus.
Substantia Nigra
The substantia nigra is generally considered part of the basal ganglia due to its similar neuronal structure and related function of motor control, but may also be considered part of the midbrain due to its location. It contains most of the neurotransmitter dopamine in the brain and also participates in motor coordination.
Diencephalon
The diencephalon is part of the forebrain and is located above the midbrain. It contains two major organs: the thalamus and the hypothalamus.
Thalamus

The thalamus is a major relay center to the cortex for all sensations except for smell. It consists of many nuclei, including the lateral geniculate nucleus, which transmits visual information, and the medial geniculate nucleus, which transmits auditory information.
Hypothalamus

The hypothalamus controls many functions including hunger, thirst, pain, pleasure and the sex drive. Another key function of the hypothalamus is to regulate the pituitary gland, which in turn, regulates hormonal levels in the body.
Medial Forebrain Bundle
The medial forebrain bundle (MFB) is a bundle of axons that passes through the hypothalamus, and is rich in dopamine neurons. When stimulated, the neurons produce reinforcing, pleasurable feelings.>
Pituitary Gland
The pituitary gland is attached to the inferior hypothalamus via a stalk containing blood vessels and neurons. The pituitary gland is divided into two distinct regions, the anterior and the posterior pituitary. The posterior pituitary is composed of neural tissue and considered an extension of the hypothalamus, while the anterior has a more extensive endocrine role, meaning that it regulates the levels of hormones in the body.
Cerebellum

At the back of the head, in between the brain stem and cerebral cortex, is the cerebellum. The cerebellum controls balance and coordination and is where learned movements are stored. Purkinje neurons that control the refinement of motor movements are found in the cerebellum. The cerebellum receives input from many parts of the brain (especially from granule cells) regarding pressure on the limbs, limb movement, and the position of the limbs in space. The dentate nucleus, located within the cerebellum, coordinates skilled movement. Damage to this region, as a result of HD, causes movements that were once smooth and refined to become jerky. Movements must also be constantly relearned.
Reticular Activating System

The reticular formation aids in regulation of the sleep-wake cycle as well as the level of arousal when awake.
Brainstem
The bottom-most part of the brain is the brain stem. The brain stem is attached to the spinal cord. It relays information between parts of the brain or between the brain and body and regulates basic body function. It is made up of the midbrain, medulla and the pons.
Midbrain: The midbrain contains the major motor supply to the muscles controlling eye movements and relays information for some visual and auditory reflexes.
Pons: The pons is a mass of nerve fibers that serves as a bridge between the medulla and midbrain above it. The pons is associated with face sensation and movement.
Medulla: The medulla (also known as the medulla oblongata) is located at the base of the brain stem and controls many of the mechanisms necessary for life, such as heartbeat, blood pressure and breathing.
Build A Brain
Now that we’ve finished our tour of the brain, let’s see how all the different sections fit together.
Credits
We hope that this tutorial has proven useful and informative to you. To leave comments or feedback, click here. Thanks for viewing the HOPES brain tutorial.
This tutorial is brought to you by:
HOPES
The Huntington’s Outreach Program for Education, at Stanford
Text by Vinita Kailasanath
Drawings by Shawn Fu
Programming & Design by Shawn Fu
Special Thanks to:
Belinda Fu, Bill Durham, Ron Barrett, and the rest of the HOPES team for their countless edits & unending patience!
Images Adapted from…
Bloom, F.E. & A. Lazerson. Brain, Mind, and Behavior. 2nd Ed. New York: W.H. Freeman, 1988.
Nieuwenhhuys, R., J. Voogd, & C. Van Huijzen. The Human Central Nervous System: A Synopsis and Atlas. 2nd Ed. Berlin: Springer-Verlag, 1981.
Nolte, J. & Jay Angevine, Jr. The Human Brain: in Photographs and Diagrams. 2nd Ed. St. Louis: Mosby, 2000.
Ornstein, R., R.F. Thompson, & D. Macaulay. The Amazing Brain. Boston: Houghton Mifflin, 1984.
Purves, W.K., D. Sadava, G.H. Orians, & H.C. Heller. Life: The Science of Biology. 6th Ed. Gordonsville: W.H. Freeman, 2001.
Sherwood, L. Human Physiology: From Cells to Systems. 4th Ed. Pacific Grove: Brooks/Cole, 2001.
Copyright 2003, HOPES
-V. Kailasanath & S. Fu, 7-15-03
More
La première apparition de la maladie de Huntington (souvent appelée “MH”) dans la littérature médicale est due au docteur George Huntington, un médecin de Long Island à New York. Cette maladie atteint aussi bien les hommes que les femmes, touchant environ une personne sur 10 000 dans la plupart des pays occidentaux. Comme les personnes atteintes de MH ont besoin de soins constants et du support de leurs proches, cette maladie fait partie de la vie de beaucoup plus de gens encore.
La ni ph
La maladie de Huntington se déclare normalement assez tardivement, quand la personne a entre 30 et 50 ans; cependant, il existe une forme de MH qui touche les enfants et les adolescents. Les personnes atteintes de MH montrent une grande variété de symptômes, que les médecins classifient habituellement en trois catégories: les symptômes moteurs, cognitifs et psychiatriques.
-
Parmi les symptômes moteurs de la MH, on peut observer spasmes musculaires, tics, rigidité, chutes, difficultés physiques à parler, et dans un état plus avancé de la maladie, difficultés à avaler (ce qui peut mener à une perte de poids significative). Des mouvements incontrôlés de torsion et de contorsion sont aussi un symptôme relativement courant de la MH. Les médecins appellent ces mouvements incontrôlés “chorée”.
-
Le principal symptôme cognitif de la MH est une modification de l’organisation des informations dans le cerveau, et en général un ralentissement du traitement de ces informations. Ces symptômes peuvent entraîner des difficultés pour apprendre des choses nouvelles, des difficultés pour s’organiser et fixer des priorités, une maladresse dans la perception de l’espace (où l’on se trouve par rapport à une table, aux murs…) et des difficultés pour porter son attention sur plusieurs choses à la fois. Il est fréquent que ces personnes se rattachent à des tâches de routine parce qu’elles leur sont plus faciles à accomplir. Enfin, à cause de troubles à organiser les mots reçus et les mots émis dans leur cerveau, beaucoup de personnes atteintes de MH ont des difficultés à communiquer avec d’autres personnes.
-
Le plus commun des symptômes psychiatriques de la MH est la dépression. Mais on peut aussi observer des troubles de personnalité, l’apathie, l’anxiété, l’irritabilité, l’obsession pour certaines activités (telles que d’aller se laver les mains), le délire et la manie. Le refus de reconnaître que l’on est atteint de MH est aussi un symptôme courant.
Malheureusement, généralement entre 10 et 25 ans après que la maladie se soit déclarée, MH a fait tellement de ravages chez la personne qu’elle en meurt, de pneumonie, crise cardiaque ou autres complications.
MH cause des détèriorations des cellules nerveuses du cerveau, entraînant des changements significatifs dans les capacités à réfléchir, ressentir et se déplacer. La cause de ces symptômes est demeurée un mystère pendant assez longtemps, jusqu’à ce que des docteurs remarquent que la maladie se retrouvait plus fréquemment dans certaines familles, et qu’ils suspectent des causes héréditaires. On sait maintenant que la transmission de la MH, comme celle d’autres traits héréditaires dépend d’informations “codées chimiquement” dans une substance appelée “acide déoxyribonucléique” ou ADN, qui existe dans les cellules vivantes. Comprendre un petit peu comment fonctionne ce code chimique permet de mieux saisir les causes de MH, et les traitements qui pourront peut-être un jour conduire à soigner la maladie.

Le code chimique de l’ADN est très similaire à la langue française: tous les deux utilisent certaines lettres dans un certain ordre pour faire passer certains messages. Mais alors que le Français a 26 lettres, l’ADN n’en a que 4: A, C, G et T (qui sont les initiales des quatre substances chimiques qui forment l’ADN). De plus, alors que la taille des mots varie beaucoup en françs, les “mots” de l’ADN sont toujours longs de trois “lettres”. Les gens qui étudient la génétique appellent ces mots des codons. Judicieusement, car les codons codent les futurs objets construits dans la cellule nerveuse. Ils sont un peu les plans, les schémas de montage. Prenons un exemple: quand un passage contient le mot anglais C-A-T (chat), vous vous représentez l’image de votre animal domestique préféré. De façon similaire, quand le code ADN contient les lettres G-G-C, il dit à la cellule de produire de la proline, un acide aminé. Pour en apprendre plus sur l’ADN, cliquer sur:
Si les codons forment les schémas de montage, alors nous pouvons voir les acide aminés qui en résultent comme des briques. Quand ces briques sont assemblées chimiquement elles forment une structure appelée protéine. Et comme dans des immeubles dans notre société, c’est dans les protéines que le travail des cellules nerveuses est effectué. Les protéines peuvent jouer des rôles très différents: elles aident la cellule à maintenir sa structure, produire de l’énergie et communiquer avec d’autres cellules.Sans les millions de protéines de notre corps; la vie telle que nous la connaissons ne pourrait pas exister.
Le comportement particulier d’une protéine est déterminé par sa forme unique dans l’espace. Cette forme contrôle comment la protéine s’imbrique et intéragit avec d’autres parties de la cellule. La forme est elle-même déterminée par les acides aminés qui composent la protéine, et par leur ordre. Et c’est comme ça qu’à la manière d’un immeuble bien conçu qui a ses origines dans les plans de l’architecte, une protéine qui fonctionne avec succès a pour origine les codons de l’ADN.
Tous les êtres humains possèdent une protéine appelée huntingtine dans leurs cellules nerveuses. (Remarquez que, bien que la “maladie d’Huntington” s’écrit avec un “o”, l’orthographe correcte de la protéine impliquée est huntingtine avec un “i”.) Les scientifiques n’ont pas encore déterminé la fonction exacte de l’huntingtine, mais elle joue visiblement un rôle critique dans les événements qui permettent aux cellules de fonctionner efficacement. Comme beaucoup d’autres protéines, l’huntingtine contient l’acide aminé glutamine. Chez les personnes atteintes de MH pourtant, il y a un nombre excessif de glutamines dans un segment de la protéine en particulier. Ces glutamines supplémentaires sont dues à un trop grand nombre de copies du codon correspondant (celui qui code “glutamine”) dans le code de l’ADN. Ce codon est le mot de trois lettres C-A-G. Il est donc exact de dire que la MH est le résultat d’un trop grand nombre de copies de C-A-G dans l’ADN qui code la protéine huntingtine. C’est pourquoi on appelle souvent la MH un “désordre de répétition trinucléotide” (“trinucléotide” est un mot savant pour “codon”.)

Combien de copies du codon C-A-G sont trop de copies? Beaucoup de recherche a été faite dans ce domaine et il y a de nombreuses réponses différentes à cette question dans la littérature scientifique. Voici une estimation grossière: les gens qui ont entre 10 et 35 copies du codon C-A-G ont une protéine huntingtine normale. Ceux qui en ont 40 ou plus, en revanche, ont une protéine huntingtine défectueuse et développeront les symptômes de la MH. En ce qui concerne les personnes qui ont entre 36 et 39 copies les choses sont moins certaines. Certains verront apparaître les symtômes alors que d’autres en seront exempts. Pour en savoir plus sur la façon dont la MH est transmise d’une génération à la suivante, cliquer ici;
Pour résumer tout ce que l’on vient de dire, la maladie de Huntington est causée par un nombre excessif de copies du codon C-A-G dans l’ADN humain, ce qui entraîne la présence de trop de glutamine dans la protéine huntingtine. Mais pourquoi l’huntingtine ainsi modifiée est-elle nuisible ? Malheureusement, et malgré les efforts vaillants des chercheurs, nous n’avons pas encore de réponse finale à cette question. Etant donné que la forme d’une protéine détermine ses interactions avec les autres parties de la cellule (comme nous l’avons appris plus haut), une importante part de la recherche sur ce point tente de comprendre exactement comment une modification de sa forme affecte les interactions de la protéine hintingtine avec les autres composants de la cellule. Une étude suggère qu’une trop grande abondance de glutamines dans la protéine huntingtine cause la formation d’agglomérats rigides de protéines. Et comme les autres composants de la cellule sont conçus pour travailler dans un environnement plus souple, ils ne peuvent plus travailler dans cette encombrement accru. Le résultat final est la mort anticipée de la cellule nerveuse (appelée apoptose). Une autre étude récente suggère que la protéine huntingtine modifiée (et plus grande qu’à la normale) “kidnape” les protéines plus petites de la cellule nerveuse, les empèchant de faire leur travail. C’est ainsi que la protéine huntingtine pourrait endommager la cellule nerveuse directement. (Pour en savoir plus sur la protéine huntigtine modifiée, click ici.)
Tandis que les scientifiques continuent à explorer les points de détail de la MH, le mécanisme de base est clair. Si nous prolongeons notre analogie avec la construction d’immeubles, ce qui se passe quand la protéine huntingtine est modifiée est que l’immeuble (la protéine) n’a ni la taille, ni la forme désirée et ne peut donc pas fonctionner correctement dans la métropole qu’est la cellule nerveuse. Et comme elle ne peut pas fonctionner correctement, elle empêche les autres protéines dont le travail dépendait du sien de fonctionner correctement. Le résultat final est un effet boule-de-neige, où les problèmes se cumulent continuellement et la cellule nerveuse devient de plus en plus endommagée. Jusqu’à ce qu’après suffisament de dommages, la cellule nerveuse en meure. Et quand ce phénomène se produit à l’échelle de nombreuses cellules nerveuses, les problèmes de réflexion, de sensation et de gestes associés à la MH peuvent être observés. Pour en savoir plus sur les cellules nerveuses et comment leur mort est liée aux symptômes de la MH, cliquer ici:
Pour en apprendre plus sur l’ADN et la MH, un grand nombre de documents sont disponibles sur le Web (ces liens pointent vers des pages anglophones, mais il existe aussi de nombreuses pages francophones) :
- Visitez Khan Academy pour un bon tutorial.
- Le site Australien “Cooperative Research Centre for Discovery of Genes for Common Human Diseases” (Gene CRC) a quelques explications fabuleuses à différents niveaux de complexité.
- Cummings, C. J and Zoghbi, H.Y. “Trinucleotide Repeats: Mechanisms and Pathophysiology.” Annu. Rev. Genomics Hum. Genet. 2000. 1:281-328.Cet article assez technique explique les symptômes de la MH, et commente le nombre de répétitions du codon C-A-G chez les personnes atteintes ou non de la maladie. Il présente aussi des théories sur le rôle de la protéine huntingtine modifiée.
- Falush D, et al. “Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial under ascertainment of late-onset cases.” Am J Hum Genet 68 (2) 2001 Feb: 373-385.Une analyse technique du nombre de C-A-G au delà duquel une personne developpera les symptômes de la MH.
- “Huntington’s Disease”. Online Mendelian Inheritance in Man. Une collection de résumés d’une multitude d’études différentes sur la MH. Cela va des études de cas sur la transmission héréditaire de la maladie aux nouvelles méthodes pour la diagnostiquer. C’est un excellent site pour se tenir au courant des différentes études et recherches menées sur la MH aujourd’hui.
- “Huntington’s Disease”. Web MD. Une présentation générale de la MH très facile à lire.
- Li SH, Lam S, Cheng AL, Li XJ. “Intranuclear huntingtin increases the expression of caspase-1 and induces apoptosis.” Human Molecular Genetics, 2000, Vol. 9, No. 19: 2859-2867. Un papier très technique expliquant comment les protéines huntingtines modifiées pourraient anticiper la mort de la cellule nerveuse (apoptose).
- Nucifora, Frederick C. Jr., et al. “Intranuclear huntingtin increases the expression of caspase-1 and induces apoptosis.” Human Molecular Genetics, 2000, Vol. 9, No. 19: 2859-2867.Un autre papier très technique expliquant comment les protéines huntingtines modifiées pourraient anticiper la mort de la cellule nerveuse (apoptose).
-M. Stenerson, 07/15/03; traduit par J.G. Morard, 11/12/04
More
La ni ar cr analog
Los Elementos Básicos de la Enfermedad de Huntington
La enfermedad de Huntington (abreviada frecuentemente “EH”) fue decrita originalmente en la literatura médica en 1872 por George Huntington, un médico de Long Island, Nueva York. La enfermedad afecta a hombres y mujeres por igual, con una incidencia de aproximadamente uno en cada 10.000 personas en la mayoría de los países occidentales. Personas con EH necesitan de cuidado especializado y el apoyo de sus familias, lo cual aumenta el número de vidas afectadas directa o indirectamente por la enfermedad.
EH es una enfermedad cerebral, degenerativa, progresiva, y hereditaria. Normalmente la edad del inicio de la enfermedad es entre 30 y 50 años, aunque hay una forma de EH que afecta a niños y adolescentes. Personas con EH pueden demostrar una gran variedad de síntomas, que los médicos agrupan usualmente en tres categorías: síntomas de movimiento, cognositivos, y psiquiátricos.
-
Algunos de los síntomas de movimiento de la EH incluyen los espasmos de los músculos, los tics, la rigidez, caídas, la dificultad para producir fisicamente el habla, y, en las fases avanzadas de la enfermedad, la dificultad para tragar (que puede causar mucha perdida de peso). También incluye movimientos ingobernables, por ejemplo torcer y retorcer el cuerpo, son síntomas ordinarios de la EH. A veces los médicos se refieren a estos movimientos ingobernables como “corea” (palabra griega que significa “baile”).
-
Los síntomas cognositivos más significativos de la EH son la organización alterada y el procesamiento lento de la información en el cerebro. Estos síntomas pueden resultar en dificultad para aprender cosas nuevas, dificultad en la planificación y toma de decisiones, el deterioro de la percepción del espacio (donde está uno en relación con las mesas, paredes, etc.), y la dificultad en hacer varias cosas simultáneamente. Las personas se adhieren frecuentemente a las rutinas ordinarias porque estas rutinas son las más faciles de aprender. Finalmente, dado que las personas con EH tienen problemas para organizar las palabras (que entran y salen) en sus cerebros, muchas personas tienen dificultad para comunicarse con otras personas a su alrededor.
-
La depresión es el síntoma más ordinario de los síntomas psiquiátricos de la EH. Otros síntomas incluyen los cambios del carácter, la apatía, la zozobra, la irritabilidad, la obsesión con ciertas actividades (como lavarse las manos), el delirio, y la manía. El rechazo a la posibilidad de tener EH es también un síntoma ordinario de la enfermedad.
Tristemente, la EH puede tomar entre 10 y 25 años después del inicio de los síntomas, en desmejorar a las personas que generalmente mueren de neumonía, insuficiencia del corazón, u otras complicaciones.
EH causa el deterioro de las células nerviosas en el cerebro (miembros de una clase de células, responsables de la señalización y funcionamiento del sistema nervioso; éstas son únicas respecto a otras células porque tienen la capacidad para comunicarse rápidamente con otras por distancias largas y con mucha precision. A veces nos referimos a las células nerviosas como neuronas.) El deterioro de estas células determina los cambios importantes que los pacientes con EH experimentan en la capacidad para pensar, sentir, y moverse. La causa de estos síntomas fue un misterio por muchos años hasta que unos médicos observaron que la enfermedad tenía “una tendencia familiar” y sospecharon así una base hereditaria (es decir, algo que es pasado genéticamente a través de varias generaciones. La herencia de EH como una enfermedad hereditaria depende en los genes que el niño recibe de sus padres.) Ahora se sabe que la herencia de la EH (así como otros rasgos hereditarios) depende en un “código químico” de información contenido en una substancia que se llama ácido desoxirribonucleico, mas conocido como ADN (la molécula de herencia; se compone de muchos subconjuntos nucleótidos arreglados en una larga cadena), que existen dentro de las células vivas. Entendiendo un poco sobre este código químico ayuda a entender mejor las causas de la EH y los tratamientos que, algún día, pueden llevar a la cura.

El código químico del ADN es semejante al idioma español: los dos usan letras específicas en un orden específico para comunicar cosas específicas. Pero mientras el idioma español tiene 29 letras, el código ADN sólo tiene cuatro A,C, G, y T (que representan los subconjuntos del ADN). Además, mientras las palabras en español pueden estar formadas por una o muchas letras, las “palabras” del ADN siempre consisten de tres letras. En la genética (que es el estudio de la herencia y de como los rasgos pasan de una generación a otra), estas “palabras” de tres letras se llaman codones. Propiamente, los codones codifican la construcción que ocurrirá en las células nerviosas. Esto es un poco semejante a los anteproyectos. Por ejemplo, cuando unpárrafo contiene las letras G-A-T-O, este pinta una figura de nuestro animal de compañía favorito. De manera semejante, cuando el código del ADN contiene las letras G-G-C, este le dice a las células que construya con prolina, uno de los veinte aminoácidos (moléculas pequeñas que son los bloques de fundación de proteínas). Para más sobre el ADN, haga clic aquí.
Si los codones son como anteproyectos, entonces podemos pensar que los aminoácidos que resultan de ellos son como unos bloques de fundación únicos. Cuando estos bloques se juntan químicamente, ellos crean la estructura de una proteína (un tipo de molécula importante en el cuerpo humano que está formado por una serie de aminoácidos. La forma de una proteína depende del número y secuencia de sus aminoácidos.). Como los edificios en una sociedad moderna, las proteínas son los sitios del trabajo de la célula nerviosa. Las proteínas tienen muchos trabajos: ellos ayudan a mantener la estructura de la célula, producir la energía, y comunicarse con otras células. Sin los millones de proteínas que tenemos en el cuerpo, la vida no podría ocurrir.

Las acciones específicas de una proteína son determinadas por su forma única de tres dimensiones. Esta forma regula como la proteína puede “encuadrar” y interactuar con otras partes de la célula. La forma es determinada por la clase de aminoácidos que compone la proteína, tanto como por su orden específico. Así como cualquier edificio que es bien construído, una proteína que funciona con buen suceso empieza con los “anteproyectos” (los codones).
Todos los humanos tienen una proteína que se llama huntingtin en sus células nerviosas. (Por favor, note que aunque “la enfermedad de Huntington” es deletreada con la letra “o”, el deletreo correcto de la proteína implicada es “huntingtin” con la letra “i”.) Aunque científicos no han determinado la funcíon exacta de huntingtin, la proteína parece desempeñar un papel crítico en los acontecimientos que ayudan a las células nerviosas a funcionar efectivamente. Como muchas otras proteínas, huntingtin contiene el aminoácido glutamina (el aminoácido clave en la EH). En las personas con EH, sin embargo, hay demasiadas glutaminas en un segmento particular de la proteína. Estas glutaminas adicionales resultan cuando una persona tiene demasiadas copias del codón correspondiente (el codón que codifica como glutamina) en el código químico del ADN. Ese codón tiene las letras C-A-G. En realidad, la EH resulta cuando una persona tiene demasiadas copias de C-A-G en el ADN que codifica como la proteína huntingtin. Por este razón, aludimos a EH como una enfermedad de la repetición de trinucleótidos (“trinucleótidos” es una palabra usada en vez de codones). Otras enfermedades de la repetición de trinucleótidos incluyen el síndrome frágil de X y la atrofia espinobulbar muscular.

¿Pero cuantas copias de C-A-G son demasiadas? Se ha realizado abundante investigación en esta área y en la literatura científica encontramos variadas opiniones en respuesta a esta pregunta. Estimaciones preliminares indican que: Personas con 10-35 copias de C-A-G tienen una forma de la proteína huntingtin que funciona normalmente. Aquellos con 40 o más copias tienen el huntingtin alterado y desarrollarán finalmente los síntomas de EH. Sin embargo, para las personas con 36-39 copias de C-A-G, el resultado no es claro. Algunas personas desarrollarán los síntomas de la EH y algunas no los desarrollarán. Para aprender más sobre la herencia de la EH, haga clic aquí.
Para resumir lo anterior, la enfermedad de Huntington es causada por demasiadas copias del codón C-A-G en el ADN, que incluye demasiadas copias de la glutamina en la proteína huntingtin. ¿Pero exactamente porque es una alteración del huntingtin perjudicial? Desafortunadamente, a pesar de los esfuerzos valientes de los científicos, nadie posee una respuesta definitiva a esta pregunta. Dado que la forma de la proteína determina sus interacciones con otras partes de la célula (tal y como fue expuesto anteriormente), mucho de la investigación a la fecha ha buscado comprender exactamente como una alteración afecta las interacciones del huntingtin con otros componentes de la célula. Un estudio indica que la superabundancia de glutaminas en el huntingtin es la causa de las agrupaciones rígidas de las proteínas. Dado que los componentes de la célula nerviosa están acostumbrados a un ambiente más flexible, no pueden funcionar con la rigidez de la proteína. Básicamente el resultado final es la muerte temprana de la célula nerviosa (a esto se le denomina apoptosis). Otro estudio reciente indica que el huntingtin alterado (y más grande que lo normal) “rapta” las proteínas menores en la célula nerviosa, previniendo su funcionamiento. De esta manera, el huntingtin alterado puede lastimar indirectamente la célula nerviosa. (Para más información sobre la proteína huntingtin alterada, haga clic aquí.)

Mientras los científicos continuan desarrollando los detalles que caracterizan la EH, el mecanismo básico es claro. Continuando con nuestra analogía de construcción del edificio, lo que pasa cuando el huntingtin está hecho en la forma alterada es que el “edificio” (la proteína) no tiene la proporción y la forma específica correcta, por lo que no puede funcionar correctamente en la “metrópoli” que es la célula nerviosa. Cuando la proteína no funciona correctamente, impide la acción de otras proteínas que dependen de la función correcta teniendo como resultado final el efecto de bola de nieve, donde los problemas que generan las proteínas continuamente danan a las células nerviosas. Finalmente, después de mucho daño, la célula nerviosa muere. Cuando muchas otras células nerviosas siguen el ejemplo, se observan los problemas de pensar, sentir, y moverse asociados con las personas que padecen EH. Para más información sobre las células nerviosas y los síntomas de EH, haga clic aquí.
Para aprender más sobre el ADN, existen muchos recursos en la red:
- Un sitio web de Australia, “Cooperative Research Centre for Discovery of Genes for Common Human Diseases” (Gene CRC) tiene excelentes guías didácticas para diferentes niveles de comprensión.
- Cummings, C. J and Zoghbi, H.Y. “Trinucleotide Repeats: Mechanisms and Pathophysiology.” Annu. Rev. Genomics Hum. Genet. 2000. 1:281-328.
Un capítulo medianamente técnico que explica los síntomas de la EH, y un desglose del número de repeticiones de CAG en las personas con y sin la enfermedad. Además, este capítulo discute las teorías con relación a la función del huntingtin alterado.
- Falush D, et al. “Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial under ascertainment of late-onset cases.” Am J Hum Genet 68 (2) 2001 Feb: 373-385.
Un análisis técnico del número de repeticiones de CAG que puede determinar si una persona desarrollará los síntomas de EH.
- “Huntington’s Disease”. Online Mendelian Inheritance in Man.
Una compilación de abstractos de una gran número de estudios de la EH. Este es un sitio excelente para toda clase de investigación de la EH. Incluye estudios de casos sobre la herencia y métodos nuevos de diagnosticar la EH.
- “Huntington’s Disease”. Web MD.
Información general sobre la EH, amistosa para los lectores. Explica generalmente la historia, los síntomas, y el tratamiento de la enfermedad.
- Li SH, Lam S, Cheng AL, Li XJ. “Intranuclear huntingtin increases the expression of caspase-1 and induces apoptosis.” Human Molecular Genetics, 2000, Vol. 9, No. 19: 2859-2867.
Una ponencia técnica sobre la manera en que el huntingtin alterado puede causar la muerte temprana (apoptosis) de la célula nerviosa.
-M. Stenerson, 07/15/03; translated by T. Altman, 11/20/04; recorded by B. Tatum 08/21/12
Podcast: Play in new window | Download
More



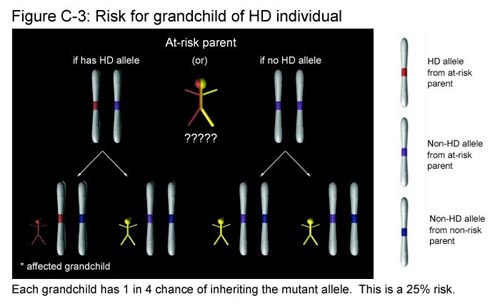













































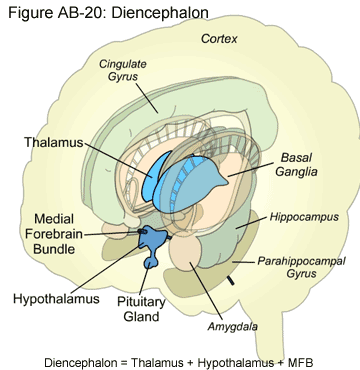



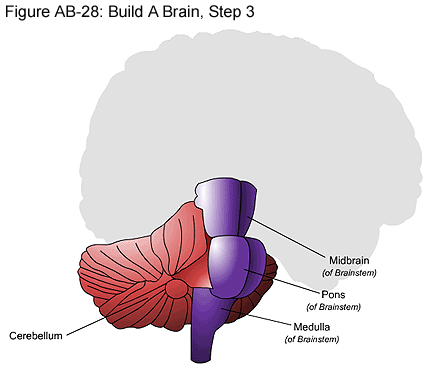











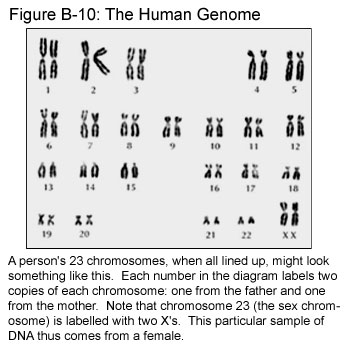{kind=link}
{kind=link}
{kind=link}