Neurotrophic factors are a family of proteins that are responsible for the growth and survival of nerve cells during development, and for the maintenance of adult nerve cells. Animal studies and test tube (in vitro) models have shown that certain neurotrophic factors are capable of making damaged nerve cells regenerate. Because of this capability, these factors represent exciting possibilities for reversing a number of devastating brain disorders, including Alzheimer’s disease, Parkinson’s disease, Lou Gehrig’s disease, and Huntington’s Disease (HD). (For more information on how HD relates to Alzheimer’s and Parkinson’s, click here.) Currently, scientists are looking for ways to harness neurotrophic factors and somehow induce the damaged nerve cells to regenerate in order to improve the symptoms of people with neurological disorders.
One neurotrophic factor that is particularly relevant to HD is Brain-derived neurotrophic factor (BDNF). BDNF levels are decreased in the brains of HD patients, which might be partly responsible for the degenerative processes of HD. Researchers have recently discovered a link between BDNF, mutant huntingtin, and excitotoxicity, a process by which brain cells die after stimulation. The mutant huntingtin protein invariably leads to the death of nerve cells in the striatum, the region of the brain needed for movements; however, how mutant huntingtin does this damage is unclear. One possibility is that mutant huntingtin lowers levels of BDNF, making nerve cells more susceptible to injury and death. Therefore, therapeutic approaches aimed at increasing BDNF production may be able to counteract the effects of mutant huntingtin and prevent a significant amount of the neurodegeneration that would otherwise occur in HD. (For more information on huntingtin protein, click here.)
What role does BDNF play in HD pathology?^
BDNF has been shown to play a role in neuroplasticity, which allows nerve cells in the brain to compensate for injury and new situations or changes in the environment. (For more information on neuroplasticity, click here.) The central nervous system (CNS) has a greater ability to recover from insult or injury than scientists had previously thought. For decades, the prevailing view was that the brain stopped developing after the first few years of life. Connections between the brain’s nerve cells could only be formed during a critical period early in life. After this critical period, it was thought that the brain was unable to form new connections. Thus, if a particular area of the adult brain was damaged or injured, nerve cells would not be able to regenerate, and the functions controlled by that area would be lost forever. However, new research suggests that this view is not entirely correct. Researchers now recognize that the brain continues to reorganize itself by forming new neural connections throughout life. Neurotrophic factors, such as BDNF, promote the survival and aid in the regeneration of adult neurons.

As mentioned above, the mutant huntingtin protein is harmful to striatal nerve cells in the brain. It also decreases transcription of BDNF, which results in a decrease in BDNF production in people who have HD. Nerve cells require BDNF to survive, but also to regenerate. Less BDNF means less neuroplasticity so the striatal nerve cells are less capable of compensating for injuries. By lowering levels of BDNF in the brain, mutant huntingtin acts as a devastating double-edged sword. First, nerve cells die because there isn’t enough BDNF to effectively combat neurodegeneration. Second, nerve cells are not able to regenerate because there still isn’t enough BDNF. It, therefore, appears that BDNF plays a crucial role in the degenerative process of HD.
How does BDNF work?^
In the brain, BDNF is released by either a nerve cell or a support cell, such as an astrocyte, and then binds to a receptor on a nearby nerve cell. (For more information on HD neurobiology, click here.) This binding results in the production of a signal which can be transported to the nucleus of the receiving nerve cell. There, it prompts the increased production of proteins associated with nerve cell survival and function.
Can exercising promote BDNF production?^
Scientists are increasingly recognizing the capacity of physical activity to maintain and compensate for the deterioration of nerve cell function. Numerous animal studies have reported that voluntary exercise leads to increased BDNF production. In rats, several days of voluntary wheel-running increased levels of BDNF in the hippocampus. This finding is surprising considering that the hippocampus is a structure normally associated with higher cognitive functions such as emotion and memory rather than motor activity. The changes in BDNF levels were found in nerve cells within days in both male and female rats and were sustained even several weeks after exercise.
Similarly, scientists studying HD in mouse models found that HD mice given the opportunity to exercise expressed more BDNF in the striatum than HD mice that didn’t exercise. This is notable because people with HD have particularly low levels of BDNF in the striatum, which is thought to be part of the reason that the striatum is the main site of neurodegeneration in people with HD. Furthermore, motor and cognitive symptoms set in later for HD mice that ran.
In order for exercise to be used as a therapeutic strategy, the type and duration of exercise would need to be determined and probably individualized to each patient. There is debate over what intensity of exercise is best to promote brain health. Although previous reports showed that only rigorous exercise, like treadmill running, stimulated BDNF expression, researchers have more recently found that even a light exercise routine may be sufficient. The downside of high intensity is that sometimes this kind of exercise can be a stressful experience that increases the release of stress hormones, thereby canceling the BDNF-promoting effects of exercise. Also, many individuals are simply unable to perform rigorous exercise. These new reports are very encouraging because they indicate that everyone can enjoy the benefits of exercise by simply engaging in light activities such as walking or doing yard work. (For more information on exercise and HD, click here).
Can BDNF be used to treat HD?^
The discovery of the relation between huntingtin and BDNF is a major step in the path to finding a treatment for HD. Previously, it was thought that mutant huntingtin gained a new function that caused neurodegeneration in the brain. However, researchers now know that HD is caused, not only by this toxic gain of function of mutant huntingtin, but also by a loss of function of normal huntingtin. Normal huntingtin allows BDNF production and plays a role in moving BDNF to the places it is needed most. In the absence of normal huntingtin, BDNF production drops drastically. This realization is a major step toward HD treatment because it indicates that therapeutics need to be aimed not only at preventing mutant huntingtin toxicity, but also at restoring normal huntingtin function.
A simple way to restore the loss of normal huntingtin function in the case of decreased BDNF production would be to administer BDNF itself. However, when BDNF is taken by routes common for other drugs, such as orally or injections into the body, it can’t reach the brain where it is needed; there is a barrier – the blood-brain barrier – that makes it difficult for substances to pass between the body and the brain. So numerous laboratories are currently trying to develop ways to deliver BDNF to the brain. However, there are still several steps that need to be taken before a drug can be developed based on this research. Scientists need to understand exactly how huntingtin “communicates” to the BDNF gene to increase its activity. Trials are already under development to deliver BDNF via gene therapy to HD transgenic mice and researchers are confident that research in this area will progress rapidly.
Research on BDNF Inducers^
While BDNF itself is not yet a viable treatment for HD, scientists are actively researching BDNF inducers, which are drugs that increase levels of BDNF in the brain.
Citalopram (Celexa)^
Citalopram is an anti-depressant that is currently on the market to treat people with depression, and goes by the brand-name Celexa. Citalopram is a particular type of anti-depressant called a selective serotonin reuptake inhibitor (SSRI). This class of anti-depressants are believed to raise BDNF levels; SSRIs cause an increase in serotonin levels, which causes nerve cells to make more BDNF. Therefore, SSRIs are being investigated for their potential ability to slow the progression of HD – as described in more detail here.
Scientists investigated how citalopram might help people with HD in a phase II clinical trial called CIT-HD. They studied the effects of citalopram on attention, thinking, muscle movements, and daily activities, with the results varying in different analyses. The study lasted for 20 weeks and concluded in 2013. For more information about CIT-HD, please click here.
Ampakines^
Ampakines are a type of drug that have recently caught the eye of the scientific community for their potential to raise BDNF levels. Cortex Pharmaceuticals Inc. is actively developing and researching the use of ampakines for treatment of various neurological disorders, including HD.
(Simmons et al. 2010): Scientists treating HD mice with ampakines are finding promising results. HD mice injected with ampakines twice a day have normal levels of BDNF. Additionally, several other symptoms of HD, such as striatal atrophy and aggregation of the mutatnt huntingtin protein, were decreased by ampakine treatment. These scientists also tested the behavior of the mice to see whether ampakine treatment was helpful in fighting the effects of the HD mutation. The motor symptoms that HD mice display were significantly improved in HD mice that were given ampakine treatment before their symptoms had begun. Another symptom that HD mice and patients display – problems with memory – seemed to be aided by ampakine treatment. Further studies are needed to verify these findings, but this study and others suggest that ampakines are a promising avenue of research.
Cystamine^
Cystamine is a drug that might combat HD in several ways. Apart from the fact that it is thought to raise levels of BDNF, cystamine might also inhibit protein aggregation (the process by which ‘clumps’ of mutant huntingtin form), and has antioxidant properties. Raptor Pharmaceuticals is currently studying cystamine in phase II clinical trials. For more information on cystamine and the on-going clinical trial, please read the HOPES article here.
For further reading^
1. Connor, J. et al. (1997). Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. Society for Neuroscience 17(7): 2295-2313.
This article is fairly complex. It describes the likely method through which BDNF exerts its effects within the brain.
2. Gomez-Pinilla, F., Ying, Z., Roy, R., Molteni, R., & V. Edgerton. (2002). Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol. 88(5): 2187-95.
This article is easy to understand and it describes the effect of exercise on brain health and plasticity.
3. Vaynman, S., Ying, Z., & F. Gomez-Pinilla. (2003). Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience 122(3): 647-57.
This article is fairly easy to read and it discusses the possible mechanisms through which exercise may influence levels of BDNF.
4. Simmons DA, Mehta RA, Lauterborn JC, Gall CM, Lynch G. Brief ampakine treatments slow the progression of Huntington’s disease phenotypes in R6/2 mice. Neurobiol Dis. 2011 Feb;41(2):436-44.
A technical article that describes how ampakines raise BDNF levels in HD mice
5. Zuccato C. et al. (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science, 293, 493-496.
This is a technical article that describes how the beneficial activity of huntingtin is lost in people with HD and how this leads to decreased production of BDNF.
6. Zuccato C., Tartari T., Crotti C., Goffredo D., Valenza M., Conti L., Cataudella T., Leavitt B. R., Hayden M. R.,Timmusk T., Rigamonti D. & Cattaneo E. (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nature Genetics 35: 76-83.
This article is very technical. It describes in detail how normal huntingtin increases transcription of BDNF by silencing NSRE.
D. McGee, 1-1-06, Updated by M. Hedlin 9.27.11 More
Verapamil is a currently available FDA-approved drug traditionally used to treat irregular heartbeats (arrhythmias) and high blood pressure by relaxing blood vessels. It has been discovered that verapamil can also modulate autophagy, a process by which cells gets rid of unwanted proteins and damaged cellular components. If this process is disrupted as it is in Huntington’s Disease (HD) and other neurodegenerative disorders then cellular “trash” can accumulate and harm brain cells. Thus, Verapamil’s effects on autophagy opens the door for its use in treating HD where the formation of protein aggregates is characteristic. To learn more about autophagy, click here. To learn more about the role of protein aggregates in HD, click here.
Verapamil was one of five L-type Ca+2 channel antagonists initially screened to test for its efficacy in modulating autophagy. L-type Ca+2 channels are specialized high-voltage ion channels found on the dendritic spines of cortical neurons. For more information about the different parts of the brain, see the brain tutorial here. Verapamil and other calcium channel inhibitors may regulate autophagy by limiting the amount of calcium that can enter the cell. High levels of intracellular Ca+2 can up-regulate autophagy by activating calpains, which are enzymes that aid in protein breakdown. Interestingly, some studies have found that calpain activity is increased in HD cells and can chop the mutant huntingtin protein into smaller fragments which allows it to enter the nucleus of neurons leading to toxicity.
Verapamil may block this toxicity by preventing calcium from entering the neuron. This lower concentration of calcium can reduce calpain activity, which can in turn increase autophagy. In HD, more autophagy means that more of the mutant huntingtin protein is cleared and fewer aggregates formed. To learn more about aggregate formation and its role in HD, click here.
The Promise of Verapamil in treating HD
Verapamil was tested for its ability to induce autophagy in several HD cellular and mouse models. To learn more about mouse models, click here. The first, and simplest model used was the cell model. Rat-derived neuronal cells were engineered to express huntingtin aggregates and some were treated with verapamil. Cells exposed to the drug showed a greater degree of aggregate clearance than cells that were not.
The next model used to explore the effects of verapamil on HD was the fruit fly, a commonly used model for many experiments. The development of the eyes in flies expressing the mutant huntingtin protein is altered, which causes the photorecetors to become disorganized and to degenerate. Flies given verapamil had less severe degeneration, than control flies did.
The next animal model of HD tested was the zebrafish. Zebrafish expressing mutant huntingtin form aggregates in their eyes and optic nerve. As in human HD, cells that form aggregates are more likely to die. Zebrafish administered verapamil had fewer aggregates.
Despite all of these experiments indicating a neuroprotective role for verapamil, the process for approving the use of verapamil in treating HD is still in very early stages. Although the results of preliminary studies are very promising, many more trials and more research needs to be done before using verapamil in HD treatments.
Further reading
- http://www.nlm.nih.gov/medlineplus/druginfo/medmaster/a684030.html
Gafni J., and L. Ellerby. “Calpain Activation in Huntington’s Disease.” Journal of Neuroscience. 2002 June; 22(12):4842-4849.
This technical paper explained how calpain activation breaks huntingtin protein into pieces small enough to enter the nucleus and lead to toxicity in HD cells.
- Sarkar S., et al. “Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies.” Cell Death and Differentiation advance online publication, 18 July 2008; doi:10.1038/cdd.2008.110.
This review paper explained the relationship between aggregate formation in several neurological diseases and the role in autophagy in protecting against these diseases. It also explained several animal models of HD.
- Williams et al. “Novel targets for Huntington’s Disease in an mTOR-independent autophagy pathway.” Nature Chemical Biology. 2008 May;5(4):295-305
This paper explained the testing of a number of potential HD drugs though targeting the autophagy mechanisms within cells.
A. Pipathsouk, 5/21/2009
More
When the cause of a disease can be traced to having too many copies of a certain nucleotide triplet in the DNA, the disease is said to be a trinucleotide repeat disorder. Today, there are 14 documented trinucleotide repeat disorders that affect human beings**. Huntington’s Disease is part of this group.
cells control nucleus refined Nis base range neuropathy
Some of these 14 trinucleotide repeat disorders are more alike than others. While the symptoms and the affected body parts vary by disease, scientists consider two illnesses to be similar if they share the same repeated codon as their cause. Six of the 14 trinucleotide repeat disorders have little or no apparent similarity to each other, or to the 8 remaining diseases. These 6 are described in brief at the end of this section. The 8 remaining disorders, one of which is Huntington’s Disease, all share the same repeated codon as their cause: CAG. Since CAG codes for an amino acid called glutamine, these 8 trinucleotide repeat disorders are collectively known as polyglutamine diseases (“poly” being the Greek word for “many”). (For background information on codons and amino acids click here.)
Polyglutamine diseases have much in common: Each of them is characterized by a progressive degeneration of nerve cells in certain parts of the body (for background info on nerve cells, click here.) In each disease, this degeneration first disrupts the function of certain group(s) of nerve cells. After 10-20 years, many of the affected nerve cells die. The major symptoms of these diseases are similar to one another and they usually affect people around the same time, in mid-life (although childhood cases have also been reported, as in the case of juvenile HD).
It deserves to be reiterated that while the polyglutamine diseases are similar to each other, they are not identical. Although they share the same repeated codon (CAG), the repeats for the different polyglutamine diseases occur on different chromosomes, and thus on entirely different segments of DNA. (For more info on chromosomes, click here.) Despite this fact, scientists are excited about research in any of the polyglutamine diseases because finding a way to stop the CAG repeat from occurring in one disease may help lead to a cure for the other 7 as well. While this is by no means a certainty, the possibility offers wonderful incentive to be persistent in research; eight for the price of one would certainly be a great deal!
Below you will find detailed descriptions for each of the polyglutamine disease, as well as a general description of all the non-polyglutamine diseases.
**Although only 14 trinucleotide repeat disorders are well-documented in medicine, genetic analysis has led researchers to believe that others exist as well. These disorders are even less common than the well-documented disorders and so have been more difficult to study, which leaves much of their story untold. They will be omitted here.
Polyglutamine Diseases:
DRPLA (Dentatorubropallidoluysian Atrophy)
Like other trinucleotide repeat disorders, DRPLA (Dentatorubropallidoluysian Atrophy) affects both the mind and body. It is characterized by abrupt muscle jerking, involuntary movements, and eventual dementia. Although these symptoms are common in the men and women of all ages who have DRPLA, young people with the disease may also be affected by progressive intellectual decline.
The Gene:
The gene involved in DRPLA lies on Chromosome 12 and is also named “DRPLA”. Typically, in asymptomatic individuals there are between 6 and 35 copies of CAG in the DRPLA allele. In a person with the disease, however, the allele has anywhere between 49 and 88 copies. At present, not enough data exist to fully understand the effect that alleles with between 35 and 49 copies of CAG will have on individuals. To learn more about alleles and more specifically, HD alleles, click here.
The Protein:
The protein product of the DRPLA gene is called atrophin-1. Although scientists are not sure about its function, the leading theory is that atrophin-1 is involved in the pathway that helps insulin take effect in the body’s cells. Since insulin helps determine how cells utilize their energy, it is essential that this pathway work smoothly so that cells can function efficiently. If there is a kink in the plan, it could spell disaster for an affected nerve cell.
How the Symptoms Come About:

The nerve cells affected in DRPLA lie in many different parts of the brain. Understanding the functions of these different parts allows us to get a better understanding of why the symptoms of DRPLA are what they are: Take first the striatum and the globus pallidus. Together, these very important regions of the brain are collectively known as the basal ganglia. The basal ganglia are important because they help plan movements and thus have a large effect on motor control. Working with other parts of the brain such as the red nucleus and the dentate nucleus (which are also damaged in people with DRPLA), the basal ganglia help to regulate each and every movement we make. When neurons in this area are damaged due to DRPLA, it’s no wonder that muscle jerks and involuntary movements become common. (For a more detailed description of the basal ganglia – written in regards to Huntington’s Disease – click here). (See Figure F-1.)

The same can be said for damage to the cerebellum, which also occurs in people with DRPLA. The cerebellum is the region of the brain where learned movements are stored. When damage occurs here, movements that were once smooth and refined become more jerky and rough since they must be constantly relearned. (See Figure F-2.)

The cerebral cortex also has a large effect on movement, particularly through the parts of it called the motor cortex and somatosensory cortex. Thus, the cerebral cortex is also involved in the motor symptoms of DRPLA. However, the tasks of the cerebral cortex reach far beyond motor control. Consider the many amazing capabilities we humans have: keen senses, the ability to speak and understand language, and the fact that we can create and use such things as logic and reason. All of these characteristics stem from functions of the cerebral cortex. Thus, when damage occurs to specific parts of the cerebral cortex, the tasks that these parts work to accomplish may become less refined. This loss of refinement may explain why people with DRPLA experience dementia when the nerve cells in the cerebral cortex are damaged. It may also partly explain the general intellectual decline in juvenile cases of DRPLA. (See Figure F-3).
Huntington’s Disease (HD)
For an introduction to HD, click here.
SBMA (Spinobulbar Muscular Atrophy)

SBMA (Spinobulbar Muscular Atrophy) occurs predominantly in males and is characterized by weakness and atrophy of the proximal muscles. Difficulties with swallowing and articulating speech are also common symptoms of SBMA. As the first word of its name implies, the disease mainly affects the spinal cord (“spino-“) and a part of the brain called the bulbar region (“-bulbar”). (See Figure F-4.)
The Gene:
The gene involved in SBMA is called the Androgen Receptor (AR) gene. It is located on the X chromosome, which is one of the so-called sex chromosomes. (Unlike most of our chromosomes, the sex chromosomes differ between males and females and this is why SBMA occurs predominantly in males. To learn more about chromosomes, click here.) Typically, in asymptomatic individuals there are between 9 and 36 copies of CAG in the AR allele. In a person with the disease, however, the allele has anywhere between 38 and 62 copies. At present, not enough data exist to fully understand the effect that alleles with 37 copies of CAG will have on individuals.
The Protein:
Hearing the oft-repeated statement “DNA codes for proteins” might lead one to believe that it is as simple as show me DNA and poof!, you have a protein. Actually, the process is much more complex than that. In fact, the full sequence of events is broken up into several parts, one of which is called transcription. The Androgen Receptor gene codes for a protein of the same name, the Androgen Receptor (also abbreviated “AR”). Because the AR protein is a key player in transcription, it is aptly titled a transcription factor.
In its normal state, then, the AR protein helps cells carry out the instructions contained within DNA. (For more in-depth discussion of DNA and the genetic code, click here.) However, in people with SBMA, the protein has extra glutamines, resulting from the extra CAGs in the AR gene. Although scientists do not yet have a definitive explanation as to why the extra glutamines cause degeneration of the neuron, it seems likely that the extra glutamines create an altered form of the AR protein that does not perform its actions in the same way as the normal AR. This mechanism for degeneration of the neuron is much like the one for Huntington’s Disease, as illustrated in Figure A-3.

Another theory suggests that the degeneration of the nerve cell is a result of neuronal inclusions (NIs). This theory, too, has its equivalent in the study of Huntington’s Disease. (Click here for more about nerve cell death in HD). According to the theory, the extra glutamines in the protein have a way of attracting other proteins to group together with the AR. This aggregation of proteins causes clumps, or inclusions, which may be solely responsible for damage to the nerve cell. More research in this area is necessary to find out definitively if the NIs are the true cause of damage. (See Figure F-5.)
How the Symptoms Come About:

Whatever the mechanism, once the nerve cells become damaged, the symptoms of SBMA begin to appear. As mentioned above, one of the main areas of the body that SBMA affects is the spinal cord. More precisely, it affects the parts of the cord known as the anterior horn and the dorsal root ganglion. The dorsal root ganglion is a group of nerve cell bodies that pass sensory information to other spinal cord nerve cells and on to the brain for analysis. The anterior horn is a region of the spinal cord that contains cell bodies of motor neurons, which put the brain’s decisions (based on the sensory info) into action. These two regions of the spinal cord are thus essential for control of fine muscle movements. When the dorsal root ganglion is damaged, the brain cannot receive proper input and thus cannot plan a movement of the muscle. When the anterior horn is damaged, the brain’s planned movement cannot be carried out. Thus, if either of these regions is not functioning correctly, then the muscles are not able to carry out the same motions that they had always done before. This inability to perform normal motions is why muscle weakness and atrophy are so common in SBMA. (See Figure F-6.)

But the spinal cord is not the only body part affected by SBMA; the bulbar region of the brain is harmed as well. The bulbar region is composed of the cerebellum, the medulla and the pons. (For a tour of brain structures, including these three, click here.) An extension of the spinal cord at the base of the brain, the medulla and pons are responsible for some of the functions that keep us alive. Functions that we usually never think about, like breathing, blood circulation, and simple actions like swallowing are all in large part controlled by the medulla and pons. More complex functions, however, require use of the cerebellum. The cerebellum is where our learned movements are stored—it helps refine a great deal of motor activities, from throwing a baseball to speaking. Given the roles of the medulla, pons, and cerebellum, it’s no wonder why damage to these areas can cause difficulty swallowing and articulating speech, two more symptoms of SBMA. (See Figure F-7.)
SCA1 (Spinocerebellar Ataxia Type 1)
SCA1 (Spinocerebellar Ataxia Type 1) is one of many closely related disorders collectively known as spinocerebellar ataxias (SCAs). Like all of the SCAs, SCA1 is characterized by atrophy of the cerebellum, a phenomenon that plays a role in the major symptoms of the disorder like loss of coordination and difficulty in articulating speech. Another common symptom of the disorder is decreased sensation in the limbs.
The Gene:
The gene involved in SCA1 lies on Chromosome 6 and is also called SCA1. Typically, in asymptomatic individuals there are between 6 and 44 copies of CAG in the SCA1 allele. In alleles with more than 20 copies (but still less than 44), the codon CAT interrupts the string of CAGs 1-4 times in a way that adds stability to the CAG chain. In a person with the disease, however, these stabilizing CATs are not present and the allele has anywhere between 39 and 81 copies of CAG. Thus, especially in the 39-44 CAG repeat range (where one may or may not be at risk for the disease), the CATs are very important—their existence can make the difference between having the illness and not.
The Protein:
The protein product of the SCA1 gene is called ataxin-1. Many studies of ataxin-1 have led scientists to believe that its major function may be to facilitate the maneuvering of nerve cell connections to allow learning. However, it is important to note that the symptoms of SCA1 are not directly caused by the loss of normal ataxin-1 function. Instead, it is believed that the cause of disease lies in the interaction between ataxin-1 and another protein called LANP. Scientists believe that LANP has a major effect on cell communication, which is needed for the survival of a nerve cell. When the ataxin-1 is altered, its interaction with LANP is also altered. The ataxin-1 is said to “sequester” the LANP and thus interfere with its normal activity. After a time, the sequestering of LANP appears to cause degeneration of the nerve cell.
How the Symptoms Come About:
To best explain how the symptoms of SCA1 come about, it is helpful to have an understanding of the cerebellum. (For more on the cerebellum, click here.)
Add to the equation a loss of pyramidal nerve cells (cells of a different pathway that are also involved in performing highly-skilled motions) and one can see why SCA1 can have such a large effect on one’s ability to perform movements.
The decreased sensation in the limbs of people with SCA1 is known as peripheral neuropathy. This condition comes about when the nerve cells that pass information from the limbs to the spinal cord (and on up to the brain) are damaged. Since they cannot do their jobs to maximum effectiveness, some of the sensory information is lost and this results in the decreased sensation.
SCA2 (Spinocerebellar Ataxia Type 2)
SCA2 (Spinocerebellar Ataxia Type 2) is characterized by a general slowing of some of the body’s normal processes. In addition to the loss of coordination that is common to all SCAs, people with SCA2 often develop slow or nonexistent reflexes and tend to shift the focus of their eyes from one point to another in a very deliberate manner. Partial paralysis of the eyes has even been described in some cases.
The Gene:
The gene involved in SCA2 lies on Chromosome 12 and is also named SCA2. Typically, in asymptomatic individuals there are between 14 and 31 copies of CAG in the SCA2 allele. In a person with the disease, however, the allele has anywhere between 36 and 64 copies. Individuals with between 31 and 36 copies of CAG may or may not develop the symptoms of the disease (individual results vary).
The Protein:

The protein product of SCA2 is called ataxin-2. So far, although the exact function of this protein is unknown, scientists believe that it may be involved in aiding protein-protein interaction within the cell. This would make it something of a “mediator” of communications within the cell. If this theory is correct, then when the protein is in its altered form in people with SCA2, it cannot do the same mediation that the normal form does. This loss of normal function means that essential protein-protein interactions cannot be as efficient as they were with the normal ataxin-2 involved. The end result is that the health of the cell is compromised. (See Figure F-9.)
How the Symptoms Come About:

The mechanism for the loss of coordination experienced in SCA2, due primarily to damage to the cerebellum, is more-or-less the same as the mechanism described for SCA1. (Read more about the cerebellum by clicking here.) The symptoms involving the eyes, however, result from SCA2’s effect on a different part of the brain. This region is called the midbrain. The primary function of the midbrain is to control the movement of the eyes. When neurons in this area are damaged, the eye’s movements become slower than normal and even partial eye paralysis can occur. Both of these phenomena are symptoms of SCA2. (See Figure F-10.)

The effect of SCA2 on the reflexes is explained by the damage it inflicts on the granule cells. A granule cell is a specific type of nerve cell that forwards a great deal of information on to the cerebellum. Much of this information involves the positions and movements of the limbs, as well as what parts of the skin are being stimulated at any given time. In terms of reflexes, all of this information is very important. As an example, suppose that someone is burned by a hot plate: The person must know not only what body part this sensation is coming from, but also where this part is located in space and what direction to move it in order to stop the pain. If this information is slow in getting to the brain, it can delay the reflex that is needed to deal with the pain. This slower flow of information occurs when the granule cells are damaged, causing people with SCA2 to develop slower reflexes. (See Figure F-11.)
SCA3 (Spinocerebellar Ataxia Type 3 or Machado-Joseph Disease)
SCA3 (Spinocerebellar Ataxia Type 3) is also known as Machado-Joseph Disease. In addition to the loss of coordination that is common to all SCAs, the most common symptoms of SCA3 include bulging eyes, small contractions of the facial muscles, and general rigidity.
The Gene:
The gene involved in SCA3 lies on Chromosome 14 and is also named SCA3 (although the name “MJD1” is sometimes used instead). Typically, in asymptomatic individuals there are between 12 and 43 copies of CAG in the SCA3 allele. In a person with the disease, however, the allele has anywhere between 56 and 86 copies. At present, not enough data exist to fully understand the effect that alleles with between 43 and 55 copies of CAG will have on individuals.
The Protein:

The protein product of SCA3 is called ataxin-3. Although scientists do not know the exact function of the protein, they do know that it normally resides in the cytoplasm of the cell. In people who have SCA3, however, ataxin-3 is known to aggregate in the nucleus. Researchers suspect that this change of place may be key in understanding the initiation of the disease. (See Figure F-12.)
How the Symptoms Come About:
Of all the polyglutamine disorders, SCA3 is perhaps the most perplexing with regard to the relationship between the affected brain regions and the symptoms of the disease. Damage commonly occurs in the cerebellum, basal ganglia, brain stem, and spinal cord. While damage to these areas commonly affects a wide range of movements, it does not seem to explain why such things as bulging eyes and general rigidity would occur. Hopefully, more research in this area will soon uncover the mystery.
SCA6 (Spinocerebellar Ataxia Type 6)
SCA6 (Spinocerebellar Ataxia Type 6) is probably the simplest of all the SCAs in terms of its symptoms: People with SCA6 predominantly experience random episodes of ataxia or slowly progressing ataxia.
The Gene:
The gene involved in SCA6 lies on Chromosome 19 and is also named SCA6. Typically, in asymptomatic individuals there are between 4 and 18 copies of CAG in the SCA6 allele. In a person with the disease, however, the allele has anywhere between 21 and 33 copies. This is the smallest number of trinucleotide repeats known to cause disease. At present, not enough data exist to fully understand the effect that alleles with 19 copies of CAG will have on individuals. Individuals with 20 copies of CAG may or may not be at risk of developing SCA6. To learn more about alleles and more specifically, HD alleles, click here.
The Protein:

Instead of its own separate protein product, the SCA6 gene codes for a subunit of the calcium channels that exist in all nerve cells. This subunit, called Alpha-1A, creates a pore in the membrane of the nerve cell, allowing calcium to enter the cell and have an excitatory effect. The excited cell can then process the inputs it has received (due to calcium’s effect) and decide whether or not it should relay this information on to other nerve cells. In this way, Alpha-1A appears to play a significant role in nerve cell communication. (For more information about how nerve cells communicate, click here). In their altered form, however, Alpha-1A subunits tend to leave the membrane and aggregate in the cytoplasm inside the cell, where they clump together and do not perform their normal duties. This movement from the membrane hinders the nerve cell’s ability to receive and process messages from other nerve cells. Since communication is essential to the survival of nerve cells, the clumping of the altered Alpha-1A subunits in the cytoplasm may play a significant role in nerve cell degeneration. (See Figure F-13.)
How the Symptoms Come About:

In SCA6, the areas most affected by nerve cell damage are the cerebellum and the Purkinje cells. Given their roles in refining motions (as mentioned in the discussion of the cerebellum), one can see how damage to these areas esults in loss of coordination. Also contributing to the symptoms is degeneration of the granule cells and the nerve cells of the inferior olive. Since these structures are involved in the input of information to the cerebellum – and likewise the Purkinje cells are involved in its output – we can see that both input and output are quite important in creating smooth, precise motions. At any given time, some nerve cells may be less affected by SCA6 than others, and this may account for the random episodes of ataxia: one group of cells may be affected one day, and another group a different day. (See Figure F-14.)
SCA7 (Spinocerebellar Ataxia Type 7)
SCA7 (Spinocerebellar Ataxia Type 7) is the last of the SCAs to fall under the category of polyglutamine diseases. Like the other SCAs, the most common symptom of SCA7 is loss of coordination. In addition to this, people with SCA7 often have difficulties with vision.
The Gene:
The gene involved in SCA7 lies on Chromosome 3 and is also named SCA7. Typically, in asymptomatic individuals there are between 4 and 19 copies of CAG in the SCA7 allele. In a person with the disease, however, the allele has anywhere between 37 and 306 copies. At present, not enough data exist to fully understand the effect that alleles with between 19 and 29 copies of CAG will have on individuals. Individuals with 30-36 copies of CAG are considered to be in the intermediate zone; they may or may not develop the symptoms of SCA7. If they do develop symptoms, the symptoms are likely to be milder and to appear later in life than they would for people with 37 or more copies of CAG.
The Protein:
The protein product of the SCA7 gene is called ataxin-7. Currently, the normal function of this protein is unknown. Scientists suspect that when ataxin-7 proteins are altered, they tend to clump together in the nucleus, producing what are called neuronal inclusions, or NIs (NIs have also been found in certain nerve cells of people with SBMA, HD, and some other SCAs). These inclusions have been associated with degeneration of the nerve cell, but whether or not they are in fact the direct cause of degeneration is yet to be determined. (See Figure F-5.)
How the Symptoms Come About:
The loss of coordination that people with SCA7 experience results from damage to the cerebellum. This mechanism is more-or-less the same as that of SCA1. (For a more detailed explanation of this mechanism, click here.)
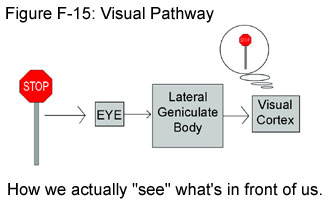
The effect that SCA7 has on one’s vision is a little more complicated because vision is a process that involves many players. Contrary to popular belief, humans do not literally “see” with their eyes. Instead, the eyes are simply the first stop on a pathway for visual information that will eventually lead to the processing of this information in the brain. After light from an image comes into the eye, the information it contains is encoded into nerve impulses by the retina. (For a discussion of nerve impulses, click here.) These impulses are then sent down the optic tract to a part of the brain called the lateral geniculate body. Here the information undergoes something like a preliminary inspection, which involves a categorization of the data. The newly categorized info is then sent on to the visual cortex, which is part of the cerebral cortex of the brain. It is in the cerebral cortex where the brain assembles a processed image and we actually “see” what is in front of us. To see an image clearly and accurately, then, all pieces in this visual puzzle must be in good working order. In SCA7, however, there is noticeable damage to all parts of the visual pathway. While this by no means implies that people with SCA7 go blind, some problems with vision are likely to occur. (See Figure F-15.)
Non-Polyglutamine Diseases
As noted in the introduction to this chapter, polyglutamine diseases are only a subset of the trinucleotide repeat disorders. As of this writing (summer 2001), researchers have identified six non-polyglutamine diseases that also fall under the category of trinucleotide repeat disorders. Because each disease involves a unique repeated codon, the six non-polyglutamine diseases show relatively little resemblance to one another. More importantly, none of them appear to have any strong similarity to Huntington’s Disease or the other polyglutamine diseases. For this reason, we provide only brief descriptions of these non-polyglutamine disorders. The descriptions follow below.
Fragile X Syndrome
Fragile X Syndrome (often abbreviated “FRAXA”) is a disorder involving the CGG codon (contrast this with the CAG codon involved in the polyglutamine diseases). The affected gene is called FMR1 and it lies on the X chromosome (hence the name “Fragile X Syndrome”). In asymptomatic individuals, the FMR1 allele has between 6 and 53 CGG repeats. In people with the disorder, the FMR1 allele has over 230 repeats. At present, not enough data exist to fully understand the effect that alleles with between 53 and 230 copies of CGG will have on individuals. Common symptoms of FRAXA include mental retardation, long and prominent ears and jaws, stereotypic hand movements (like flapping and biting one’s hands), hyperactivity, and others. The disease typically affects males.
Fragile XE Mental Retardation
Fragile XE Mental Retardation (often abbreviated “FRAXE”) is a disorder involving the GCC codon. The affected gene is called FMR2 and, like the gene causing Fragile X Syndrome, FMR2 lies on the X chromosome. In asymptomatic individuals, the FMR2 allele has between 6 and 35 copies of GCC. In people with the disorder, however, the allele has over 200 copies of GCC. At present, not enough data exist to fully understand the effect that alleles with between 35 and 200 copies of GCC will have on individuals. Common symptoms of FRAXE include mild mental retardation, learning deficits, and possible developmental delays.
Friedreich’s Ataxia
Friedreich’s Ataxia (often abbreviated “FRDA”) is a disorder involving the GAA codon. The affected gene is called X25 (also known as “frataxin”). In asymptomatic individuals, the frataxin allele has between 7 and 34 GAA repeats. In people with the disorder, the allele has 100 or more repeats. At present, not enough data exist to fully understand the effect that alleles with between 34 and 100 copies of GAA will have on individuals. There are many common symptoms of FRDA, some of which include slurred speech, heart disease, and diminished reflexes of the tendons. The name “ataxia” describes a loss of coordination, and this is typical in the limbs and trunk of those who have FRDA. The typical age of onset for this disorder is early childhood.
Myotonic Dystrophy
Myotonic Dystrophy (often abbreviated “DM”, not “MD”) is a disorder involving the CTG codon. The affected gene is called DMPK. In asymptomatic individuals, the DMPK allele has between 5 and 37 CTG repeats. In people with the disorder, the allele has at least 50 repeats in adult-onset cases, and can go up to several thousand in congenital cases. At present, not enough data exist to fully understand the effect that alleles with between 37 and 50 copies of CTG will have on individuals. Common symptoms of adult-onset DM include muscle weakness and degeneration, while such symptoms as kidney failure, facial dysmorphology, heart problems, premature balding, cataracts, and, in males, atrophy of the testicles are less common. The congenital form of DM is the most severe and its symptoms include diminished muscle tone, problems with respiration at birth, and developmental abnormalities. The term “myotonic” comes from “myotonia”—a condition characterized by frequent muscle spasms. Obviously, myotonia is quite common in DM.
Spinocerebellar Ataxia Type 8
Like Myotonic Dystrophy, SCA8 (Spinocerebellar Ataxia Type 8) is a disorder involving the CTG codon. The affected gene is also called SCA8. Asymptomatic individuals possess between 16 and 37 repeats of CTG in the SCA8 allele, while people with the disorder have between 110 and 250 repeats. At present, not enough data exist to fully understand the effect that alleles with between 37 and 110 copies of CTG will have on individuals. SCA8 is a slowly progressive disorder and its symptoms include decreased sense of vibration, sharp reflexes, and atrophy of the cerebellum, which has a large amount of control over the body’s learned movements. (For a more detailed description of the cerebellum, click here.)
Spinocerebellar Ataxia Type 12
Much like the polyglutamine diseases discussed above, SCA12 (Spinocerebellar Ataxia Type 12) is a disorder involving the CAG codon. But unlike the polyglutamine diseases, which have CAG repeats that occur in what is known as the “translated region” of DNA, the CAG repeats in SCA12 occur in what is called an “untranslated region” of DNA. In what basically amounts to an exception to the normal rule, the chemical information of an untranslated region of DNA is not used as instructions for making proteins. None of the codons in the untranslated region of DNA produce any amino acids at all (a realization that has prompted some scientists to refer to the untranslated region as “junk DNA”). This exception means that the CAG codons of SCA12 actually do not produce the amino acid called glutamine. Because of this fact, SCA12 is not considered a polyglutamine disorder.
The affected gene in SCA12 is also called SCA12. Asymptomatic individuals possess between 7 and 28 repeats of CAG in the SCA12 allele, while people with the disorder have between 66 and 78 repeats. At present, not enough data exist to fully understand the effect that alleles with between 28 and 66 copies of CAG will have on individuals. SCA12 is the most recent addition to the group of spinocerebellar ataxias. Since there are relatively few cases to date, the full effects of the disorder are not yet fully known. Given that it is a spinocerebellar ataxia, however, it is likely that some of the general symptoms include slurred speech and loss of coordination of some parts of the body.
For further reading
- Cummings, C. J. and Zoghbi, H.Y. “Trinucleotide Repeats: Mechanisms and Pathophysiology.” Annu. Rev. Genomics Hum. Genet. 2000. 1:281-328.
A fairly technical paper explaining the symptoms of each trinucleotide repeat disorder, as well as a breakdown of the codon involved and the amount of repeats in people with and without the disease (as of the publishing, however, updated and slightly different data regarding the numbers are available; see next entry in bibliography). Also discussed are theories regarding the function of the altered proteins.
- GeneClinics. Online.
An in-depth site with very recent information about all of the SCAs (and DRPLA). A wonderful resource to find out more about each disorder. (Look up the any of the SCA’s by using the search feature.)
- Online Mendelian Inheritance in Man (OMIM). Online.
A compilation of abstracts from a multitude of different studies on HD. From case studies regarding inheritance to new methods of diagnosing HD, this is an excellent site for all the various types of HD research going on today. (Look up any of the trinucleotide repeat disorders using the search feature.)
- Silverthorn, Dee Unglaub. “Human Physiology.” Upper Saddle River, NJ: Prentice Hall, 2001. pp. 256-263, 396.
Written for college students, this textbook has excellent explanations of all aspects of human physiology, as well as wonderful pictures to increase one’s understanding. The pages noted are excellent in teaching the functions of various parts of the nervous system.
- Thompson, Richard F. “The Brain.” New York: Worth Publishers, 2000. pp. 11-16, 296-303, 308-309, 451.
An introduction to neuroscience. Very clearly explains the functions of the various parts of the nervous system. Also gives insight into current research going on in neuroscience.
-M. Stenerson, 9-25-01
More
In the last few years, stem cell research has become the latest buzz in the popular media as well as the scientific world. It was the subject of President George W. Bush’s first prime-time television address. It is continuously on the cover of popular news magazines. So what is all the fuss about?
Stem cells hold the potential to treat or even cure many of the diseases that continue to mystify scientists today, such as Parkinson’s Disease, Alzheimer’s Disease, diabetes, and Huntington’s Disease (HD). However, stem cell research is controversial, as most of the stem cell lines available today are derived from embryos or fetuses.
basic ups
The following chapter aims to explain the science behind stem cells and their potential to treat HD.
Stem Cell Basics
What is a stem cell?
Most of the cells that make up the organs and tissues of the body are highly specialized for their specific jobs. The red blood cell, for example, is specifically crafted to carry oxygen from the lungs to the tissues. A comparison can be made with today’s society where most workers are trained to perform a specific trade. The days of the generic fix-it man are gone; instead, the electrician, the plumber, and the cable guy fill specific niches.
Likewise, in the human body, most cells are specialized for certain jobs. In fact, most cells lead very standard lives – they grow up, do the same job every day, and then eventually retire and pass away. These cells, such as nerve cells or skin cells, are called specialized cells. They are mature cells that have characteristic shapes and are committed to performing specific functions (See Figure 1). Once these cells have matured, they are usually incapable of reproducing themselves. They essentially remain “childless” for their whole lives.
If mature specialized cells cannot leave “children” behind when they die, how does your body make new cells? For example, when you cut your skin, how do you grow new skin cells? When you get blood drawn, how do you make new blood cells?
It turns out that stem cells solve this unique problem. A stem cell can reproduce itself over and over again (a special trick known as “self-renewal” or “self-replication”). With every replication, the stem cell produces one new stem cell and one new specialized cell. Stem cells can often give rise to a number of different cell types. For example, blood stem cells can produce both red blood cells and white blood cells. In this way, stem cells are not committed to produce a single cell type. Instead, a stem cell remains uncommitted until it receives a specific signal to divide and produce one of the various specialized cells.
In more formal terms, a stem cell is a special kind of cell that has the ability to divide for indefinite periods of time and to give rise to the mature, specialized cells that make up an organism. A stem cell is uncommitted and remains uncommitted until it receives a signal to differentiate (become a specialized cell). (See Figure 2).
What are the different kinds of stem cells?
There are three main types of stem cells under scientific study today:
- Embryonic stem (ES) cells: ES cells are taken from the very early stages of embryo development and can give rise to all of the cells of the human body, except the placenta and other supportive tissues in the womb.
- Embryonic germ (EG) cells: EG cells are taken from the later stages of embryo development and are slightly less “powerful” in their ability to divide.
- Adult stem cells: Adult stem cells are found in the tissues of a fully developed child or adult and can only produce a limited number of cell types.
These three types of stem cells are easiest to understand in a discussion of human development. Human development begins when a sperm fertilizes an egg and creates a single cell, known as a zygote, which has the potential to form an entire organism. This single cell is said to be totipotent, meaning it has the “total” potential to give rise to all types of cells. About 24 hours after fertilization, the zygote divides into two identical totipotent cells, and is now known as an embryo. About five days after fertilization and after several cycles of cell division, these cells begin to specialize and form a hollow sphere, called a blastocyst. The blastocyst has an outer layer of cells that make up the shape of a sphere and a cluster of cells, known as the inner cell mass, inside the sphere. The outer layer of cells will eventually form the placenta. The inner cell mass will eventually form all the tissues of the human body. The inner cell mass cannot form an organism on its own, however, because it is unable to produce the placenta and the other supporting tissues necessary for development in a woman’s uterus. Therefore, the inner cell mass cells are said to be pluripotent, meaning they have the potential to give rise to most of the tissues required to produce an organism. In other words, they can give rise to all the cells of the human body, excluding the supportive tissues used in the womb. (See Figure 3).
Embryonic stem cells, which are also pluripotent, are isolated directly from the inner cell mass at this blastocyst stage. In 1998, researchers first isolated ES cells from human embryos that were obtained from in vitro fertilization clinics. Although these embryos were originally intended for reproduction, they were in “excess” and were headed for the trash. Instead of being disposed, however, they were donated to research.
Five to 10 weeks after fertilization, the growing embryo, now called a fetus, develops a region known as the gonadal ridge. The gonadal ridge contains the primordial germ cells, which will eventually develop into eggs or sperm.
Embryonic germ cells are isolated from these primordial germ cells of the 5- to 10- week old fetus. Like ES cells, EG cells are also pluripotent.
As the human fetus continues to develop, pluripotent stem cells specialize into stem cells that are geared for specific tissues. For example, they become blood stem cells (which produce blood cells) or skin stem cells (which produce skin cells). These specialized stem cells are said to be multipotent, meaning they can give rise to many, but not all, types of cells.
While all three types of stem cells discussed above (ES cells, EG cells, and multipotent stem cells) are found in the developing human, only multipotent stem cells are found in children and adults. Therefore, multipotent stem cells are often referred to as adult stem cells. Unlike other stem cells, adult stem cells are only found in specialized tissues and can only give rise to the specialized cell types that make up that tissue. Currently, adult stem cells have been found in the bone marrow, blood, blood vessels, skeletal muscle, skin, lining of the digestive track, dental pulp of the tooth, liver, pancreas, cornea and retina of the eye, and brain.
Stem Cell Research
What kind of research is being conducted?
Stem cells are being investigated in various areas of scientific research. The most notable research areas are described below:
I. Basic Research
On the most fundamental level, stem cells are used to study the early events of human development. This research may one day explain the cause of birth defects and help devise new approaches to correct or prevent them. Also, research on the genes and chemicals that control human development may help researchers manipulate stem cells to become specialized for transplantation or genetic engineering.
II. Transplantation Research
Stem cells may hold the key to restoring many vital bodily functions by replacing cells lost in various devastating diseases. Many diseases and disorders, such as Huntington’s disease, disrupt specific cellular functions or destroy certain tissues in the body. The goal, therefore, is to coax stem cells to develop into the desired specialized cells, which can then be used as a renewable source of replacement cells or tissues. This process could possibly treat HD and other conditions such as Parkinson’s and Alzheimer’s diseases, spinal cord injury, stroke, burns, heart disease, diabetes, osteoarthritis, and rheumatoid arthritis.
III. Genetic Engineering Research
Stem cells could be used as a vehicle for delivering genes to specific tissues in the body. The goal is to add genes to stem cells that would then coax the stem cell to differentiate into a specific cell type or force the stem cell to produce a desired protein product. Currently, researchers are trying to use specialized cells derived from stem cells to target specific cancer cells and directly deliver treatments that could destroy them.
IV. Drug Testing and Toxin Screening
Currently, animal models are used to test drug safety and efficacy and to screen potential toxins. Animal models, however, cannot always predict the effects that a drug or toxin may have on human cells. Therefore, if human stem cells can be used to generate cells that are important for certain drug or toxin screenings, these cells may offer a safer, more reliable test by mimicking a more realistic human environment.
V. Chromosomal Abnormality Testing
Stem cells might also be used to explore the effects of chromosomal abnormalities in early human development. As a result, we might be able to understand and monitor the development of early childhood tumors, many of which are embryonic in origin.
What are the advantages and disadvantages of using embryonic stem cells, embryonic germ cells, and adult stem cells for research?
At first glance, embryonic stem cells, embryonic germ cells, and adult stem cells all present similar possibilities for scientific research. They are all stem cells, after all, and therefore share some key characteristics and hold similar potential. For example, they all have the ability to self-replicate for indefinite periods of time in the human body and can give rise to specialized cells. The overall purpose behind research with all types of stemm cells, therefore, is very similar. It has also been shown that all three cell types can be isolated from other cells and kept in a specific laboratory environment that keeps them unspecialized. This is crucial for controlled scientific research. Upon experimentation, it has also been shown that all stem cell types will replicate and specialize when transplanted into an animal with a lowered immune system. The cells then undergo “homing,” a process where the transplanted cells are attracted to and travel to an injured site when transplanted into an animal that has been injured or diseased. Homing provides hope that the transplantation of stem cells will be a clinically useful procedure.
Despite these general similarities, there are some important differences between embryonic stem cells, embryonic germ cells, and adult stem cells. The origins of these three cell types define their differences: ES cells are derived from the inner cell mass of the blastocyst in a developing embryo, EG cells are obtained from the primordial germ cells of a fetus, and adult stem cells are found in developed, specialized tissues. The differences between ES, EG, and adult stem cells result in different advantages and disadvantages for each stem cell type in scientific research and development.
ES and EG cells have some clear advantages over adult stem cells concerning research and clinical usefulness. For example, ES and EG cells are pluripotent, meaning they have the potential to give rise to all types of cells in the body. Adult stem cells are multipotent, meaning they only have the potential to give rise to a limited number of cell types. So far, no adult stem cells have proven to be pluripotent. This means that ES and EG cells could potentially provide a renewable source of replacement cells for any tissue in the human body. Adult stem cells, however, would only be clinically useful for the specific adult tissue that the stem cells came from. ES and EG cells are also relatively abundant in the developing organism, especially compared to adult stem cells, which are scarce in the adult body. As a result, ES and EG cells are much easier to identify, isolate, and purify compared to adult stem cells, which are very difficult to identify, isolate and purify in the lab. This makes research with ES and EG cells all around easier than research with adult stem cells.
On the flip side, adult stem cells have some distinct advantages over ES and EG cells. For example, adult stem cells are around for an organism’s lifetime, while ES and EG cells are only found in the developing organism. This allows a longer time frame for adult stem cells to be studied in an individual. Also, removal of stem cells from an embryo will result in the death of the embryo. Removal of adult stem cells, however, does not involve the death of an embryo, and is therefore less ethically complicated. Furthermore, adult stem cells pose no chance of immune rejection after transplantation because they can be transplanted back into the adult that they came from. ES and EG cells are derived from embryos and fetuses, however, and are transplanted into people with different genetic make-ups. Therefore, rejection is an issue only with the use of ES and EG cells.
Finally, ES cells have a strong advantage and disadvantage over the other stem cell types. First, ES cells are able to replicate in the laboratory far better than either EG or adult stem cells. ES cells can self-renew for up to 2 years, doubling up to 300 times. EG cells can only double a maximum of 70-80 times. Meanwhile, adult stem cells only have a limited ability to replicate in the lab. Replication in the laboratory is critical for research to continue. On the other hand, ES cells are the most likely to develop into tumors. If undifferentiated ES cells are taken from the lab and injected into a mouse, a benign tumor can develop. For this reason, scientists do not plan to use undifferentiated ES cells for transplants or other therapeutic applications. EG cells do not form these tumors, however. At this point, it is not known whether tumors will form with transplanted adult stem cells.
The similarities and differences of ES, EG, and adult stem cells are summarized in the chart below:
Which are more useful – pluripotent stem cells or adult stem cells?
Based on what scientists currently know, it is unclear whether pluripotent or adult stem cells will be more useful for the development of therapies. As far as scientists can tell at this point, neither one is probably better than the other.
Both pluripotent and adult stem cells have their advantages and disadvantages (see chart below). For example, the main advantage of pluripotent stem cells is their ability to produce any specialized cell in the human body. However, because they are derived from human embryos or fetuses, they are also very controversial.
Adult stem cells, on the other hand, are unlikely to be rejected by a patient’s immune system because they can be isolated from a patient, coaxed to divide and specialize, and then transplanted back into the patient. Because stem cells are isolated from an adult, they are also unlikely to cause ethical concerns. However, adult stem cells have not been isolated for all tissues of the body, which limits the types of tissues they can be used for.
Recently, there has been research on adult stem cell plasticity, the ability of an adult stem cell from one tissue to generate specialized cells of another tissue. Thus far, there have been contradicting results. Time will tell whether or not adult stem cells can actually demonstrate plasticity. For more information on cell plasticity, click here.
Many scientists agree that pluripotent and adult stem cells might be better suited for different treatments
What challenges are researchers facing?
While stem cell research shows great promise, researchers continue to face many biological, technological, and ethical challenges that must be overcome before innovations can be developed and incorporated into clinical practice.
First, more basic research must be done in order to fully understand the events that lead to cell specialization in humans. Currently, scientists are working to produce reliable, reproducible conditions that will direct stem cells to become the specific types of cells and tissues that are needed for transplantation.
Also, before mature cells derived from ES or EG stem cells can be used for transplantation, scientists must overcome the problem of immune rejection. Because these cells are genetically different from the recipient, their incompatibility must be minimized.
Adult stem cell research has also faced many difficulties, including finding, isolating and identifying the cells, growing the adult stem cells in the laboratory and demonstrating plasticity.
In addition to these technological challenges, researchers must also face the ethical controversy surrounding the use of ES and EG cells. If stem cells are used in clinical practice, researchers, doctors, and society at large must agree on acceptable ethical guidelines.
Stem Cell Research and Huntington’s Disease
What is the potential for using stem cells to treat HD?
Huntington’s Disease is a neurodegenerative disorder that is characterized by the death of nerve cells in the striatum. (To learn more about the neurobiology behind HD, click here.) Until recently, it was believed that neurons in the adult human brain and spinal cord could not regenerate. Once dead, the neurons were thought to be gone for good. In the mid-1990s, however, researchers discovered that stem cells in the adult brain could give rise to new neurons and neural support cells. With their ability to regenerate and produce new nerve cells, neural stem cells might be able to replace or repair the cells that are destroyed by HD, thus restoring lost function.
In fact, researchers have already discovered how to coax embryonic and adult mouse stem cells to develop into neurons that produce a neurotransmitter called gamma-aminobutyric acid – the type of neurons that are mainly lost in HD.
More research could potentially lead to the following:
- If these stem cells can produce nerve cells in the laboratory, they could be transplanted into the striatum to replace the lost nerve cells, or;
- If the adult stem cells already present in the patient’s brain could be stimulated to produce more neurons, they might be able to “self-repair” the striatum.
Either way, further stem cell research could yield new treatments to HD given enough time, research, and luck.
What is fetal neural transplantation? What does this have to do with HD and stem cells?
Fetal neural transplantation is a surgical technique that involves removing nerve cells from an aborted fetus and transplanting them into a human patient. Clinical trials have attempted to use this technique as a treatment for HD by removing striatal nerve cells (those mainly affected by HD) from a human fetus and grafting them into the brain of an adult patient. The therapeutic value of fetal transplantation has been promising so far. Notable improvements include increases in brain activity and motor and cognitive functions. Although the initial results have been encouraging, the clinical usefulness of fetal neural transplantation for HD treatment remains unclear.
The use of human fetal tissue creates a major roadblock to the development of this technique for two reasons. Technically, fetal tissue is difficult to obtain and prepare. Ethically, the use of fetal tissue raises serious concerns. Therefore, the development of an alternative source of nerve cells for neural grafting will be crucial for the continuation of neural transplantation research. Stem cells currently hold great potential as an alternative source. Theoretically, neural stem cells could be developed in the laboratory and then grafted into the patient’s brain. Ultimately, the future of fetal neural transplantation as a clinically effective HD therapy relies heavily on the future of stem cell research.
Will stem cell research provide the cure for HD?
Researchers generally do not believe that stem cell research will be the “magic cure” for HD. Rather, it is likely to be part of the fight against the neurodegeneration seen in HD. Ultimately, the medical and scientific community will need to improve early diagnosis, reduce the severity of cell loss, combat inflammation, provide new neurons (which is where stem cells factor in), and utilize progressive rehabilitation techniques to allow complete regeneration. While stem cells may not cure HD, they could serve as a crucial component to effective treatment.
For further reading
- Allison, Wes. “Preliminary success of fetal brain-cell transplantation in Huntington’s Disease.” The Lancet.
A short, but fairly technical article.
- Begley, Sharon. “Cellular Divide.” Newsweek, 9 July 2001: 22-27.
An easy-to-read explanation of stem cells and an update on progress as of July 2001.
- Bjorklund, Anders and Ollie Lindvall. “Cell replacement therapies for central nervous system disorders.” Nature Neuroscience, June 2000, 3 (6): 537-544.
A technical paper discussing the progress of fetal neural transplantation in treating Parkinson’s and Huntington’s Disease.
- Freeman, Thomas, et.al. “Tranplanted fetal striatum in Huntington’s disease: Phenotypic development and lack of pathology.” Proceedings of the National Academy of Sciences of the United States of America, 5 December 2000, 97 (25): 13877-13882.
A highly technical paper discussing the potential of fetal neural tissue to treat HD.
- Gibbs, W. Wayt. “Biological Alchemy.” Scientific American, February 2001: 16-17.
A less technical article depicting the discovery of neural adult stem cells and discussing the possible plasticity of adult stem cells.
- Mitchell, Steve. “Rare stem cells produces many cell types.” United Press International, 21 June 2002.
A short, easy-to-read article about adult stem cell plasticity.
- “Stem Cells: A Primer.” National Institutes of Health, May 2000.
A comprehensive, easy-to-read explanation of stem cells and their potential applications. Great online resource.
- “Stem Cells: Potential for Good?” The Economist, 18 August 2001: 59-61.
A thorough explanation of stem cells and the controversy surrounding their development and use.
- “Stem Cells: Scientific Progress and Future Research Directions.” Department of Health and Human Services, June 2001.
An extensive, fairly technical summary of everything you would want to know about stem cells.
- Weiss, Samuel. “Stem Cells and Huntington Disease.” Horizon, Huntington Society of Canada Newsletter, Summer 2001, No. 101: 1-2.
An easy-to-read explanation of stem cells and their potential to treat HD.
-J. Czaja, 3-07-03
More
Readers of our website sometimes ask, “Where does all the research summarized by HOPES come from?” Here follows a list of some main contributors to HD research, along with some of their recent studies, clustered in five categories:
We’ve also included some news articles highlighting the key discoveries. You will notice that the “Recommended Reading” list at the end draws directly from medical journals, so please be forewarned: the material there may be complex.
Ph
Biological Basis of HD
Johns Hopkins University School of Medicine
http://www.hopkinsmedicine.org/psychiatry/specialty_areas/huntingtons_disease/index.html
Director: Christopher A. Ross, M.D., Ph.D.
- Discovered a gene that, when mutated, causes a disorder called “Huntington’s disease-like 2,” or HDL2, which is very similar to HD.
- Found a way to make the CREB binding protein (a protein involved in the neuronal effects of HD) harmless by cutting out certain molecular areas.
- Determined that HD causes movement abnormalities by preventing the basal ganglia in the brain from correcting its mistakes.
Representative News Article:
http://www.jhu.edu/~gazette/2002/28oct02/28novel.html
University of California, Irvine
- Found that a certain protein called arfaptin 2 can prevent the huntingtin protein from aggregating, although it is still unclear whether or not these aggregates cause HD.
- Discovered that a class of drugs called “histone deacetylase inhibitors” can actually prevent, and in some cases reverse, brain cell death in fruit flies.
Representative News Article:
https://news.uci.edu/briefs/thompson-studies-point-to-new-targets-for-huntingtons-disease-treatments/
Weill College of Medicine at Cornell University
- Used metabolism studies to show that mice who have the HD allele do not use energy as efficiently as normal mice do.
University of Pennsylvania School of Medicine
- Used fruit flies to show that certain protein chaperones may suppress HD.
- First to discover that HD causes proteins to aggregate in the brain.
Stanford University
- Found that huntingtin aggregates cause proteasomes to malfunction, which creates problems when toxic proteins collect.
- For more information about research at Stanford, click here.
Mayo Clinic
- Researched why mutant proteins cannot be degraded in patients with HD.
HD Genetics
Indiana University School of Medicine
- Sends researchers to Lake Maracaibo, Venezuela, a community that has a high number of related individuals who are predisposed to developing HD. Researchers obtain specimen samples and test neurological functions to determine the genetic inheritance of HD.
University of Southern California
- Studied the Lake Maracaibo community and found that HD is caused by many genetic mutations that happen during mitosis—not just one in meiosis—and that the number of CAG repeats on the HD allele increases as the mutations accumulate over time.
Massachusetts General Hospital
- Found that certain types of brain cells tend to appear in higher densities in people who have a family history of HD.
- Studied the effects of HD on the transcription of genes across generations.
The Search for Treatments
Massachusetts General Hospital
- Tested coenzyme Q10 as a potential treatment for HD, and found that the drug can extend patients’ lives and delay the onset of symptoms.
- Performed an inconclusive clinical trial of riluzole, a drug that had been shown to improve motor abnormalities in HD-afflicted baboons. Investigations of riluzole continue.
National Institutes of Aging (NIA)
- Recently found that periods of fasting decreased the symptoms of HD in mice. Low-calorie diets and reduced meal frequency can both delay the onset of HD and slow down the spread of the disease.
Representative article:
http://www.sciencedaily.com/releases/2003/02/030211072836.htm
University of South Florida Center for Aging and Brain Repair
- Found that people whose diets are rich in antioxidants age slower because antioxidants block the free radicals that cause body function to decline.
- Successfully slowed aging in the brain by implanting stem cells from human umbilical cord blood.
- Discovered that when fetal tissue is implanted in the brains of HD patients, the new tissue remains free of HD.
Representative articles:
http://www.mcleanhospital.org/PublicAffairs/20001130_huntingtons.htm
http://www.cnn.com/2001/fyi/news/08/09/fetal.cell/
Stanford University
- Challenged the relationship between protein aggregates and HD with the discovery that drugs such as cystamine can reduce the symptoms of HD without affecting aggregates.
Weill College of Medicine at Cornell University
- Conducted key research on potential HD treatments such as coenzyme Q10 and creatine.
Psychological Studies
Columbia Health Science HDSA Center for Excellence at the New York State Psychiatric Institute
- Found that HD patients’ grasping abilities decrease more and more over time.
- Studied a group of HD patients and found that over half of them had symptoms of obsessive-compulsive disorder.
- Currently exploring the safety of creatine for the treatment of HD.
Indiana University School of Medicine
- Recruited patients with a family history of HD and found that the ones who eventually developed the disease became more irritable and hostile before any other signs of HD appeared.
Johns Hopkins University School of Medicine
- Most HD patients also suffer from depression, impaired thinking, personality changes, and other disorders that can be treated with medication.
University of Connecticut Medical School
- Found that when HD patients are asked to do a cognitive task (like solving a word puzzle, for instance), less blood flows through their brains than through the brains of people who don’t have the disease.
University of Pennsylvania School of Medicine
- Found that HD patients are less able to identify odors than people who do not have HD.
Link to Publication:
http://www.med.upenn.edu/stc/papers/1997/97byla.pdf
Clinical Trials
Huntington Study Group (HSG)
HSG is a worldwide collaboration of researchers and physicians who work together to study HD. The group has produced numerous studies in the past; current projects include:
- MINO-HD: Aims to determine exactly how the drug minocycline affects HD patients. There is already evidence suggesting that the drug has positive effects, since minocycline prevents the expression of caspase, an enzyme that triggers certain events that lead to cell death.
- Prospective Huntington At Risk Observational Study (PHAROS): Plans to monitor people who are genetically at-risk for developing HD and to try and determine the circumstances under which these people actually go on to develop the disease.
- PREDICT-HD: Compares the brains of HD patients with those who are “at-risk” for HD to find out what triggers the disease. The difference between this study and PHAROS is that PREDICT-HD is recruiting patients who already know that they have the HD allele, whereas volunteers for PHAROS cannot know whether or not they have the mutation.
University of Rochester School of Medicine and Dentistry
- Researched the rate at which HD patients’ motor skills decline.
- Developed a method to test the reliability of diagnosing HD.
For Further Reading:
- Albin RL. Fetal striatal transplantation in Huntington’s disease: time for a pause. Journal of Neurology, Neurosurgery, Psychiatry. 2002 Dec; 73(6):612.
- Bonini NM. Chaperoning brain degradation. Proceedings of the National Academy of Sciences. Dec 10;99 Suppl 4:16407-11.
- Cattaneo E, Rigamonti D, Zuccato C. The enigma of Huntington’s disease. Scientific American. 2002 Dec; 287(6): 92-7.
- Kieburtz K. Issues in transplantation for Huntington’s disease. Cell Transplantation. 1999 July/Aug;8:456-457.
- McMurray CT. Huntington’s disease: new hope for therapeutics. Trends in Neuroscience. 2001 Nov;24(11 Suppl):S32-8.
- Parker M, Lucassen A. Working towards ethical management of genetic testing. Lancet. 2002 Nov 23;360(9346):1685-8.
- Sharma N, Standaert DG. Inherited movement disorders. Neurological Clinician. 2002 Aug;20(3): 759-78.
More
When it comes to scientific research, the public wants results and we want them fast. This is especially true of research on chronic or fatal human diseases such as diabetes, cancer, and Parkinson’s, which affect millions of people in the United States alone. Because the public loves good news, the media is quick to report stories involving major scientific breakthroughs (or what appear to be). On June 16, 2006, for example, the Canadian press released an article entitled “Canadians cure Huntington’s disease in modified mice.”
As I learned firsthand this summer as an intern at Dr. Marcy MacDonald’s Huntington’s disease (HD) laboratory in the Center for Human Genetic Research at Massachusetts General Hospital (MGH) in Boston, the disease is far from cured, even in mice. In fact, the research community is still years, perhaps decades, away from finding drug treatments that target the genetic mutation whose deleterious effects lead to HD, a neurological disorder with symptoms that typically begin in middle age. HD is termed “neurodegenerative” because it involves a progressive loss of nerve cells in the brain. The disease affects men and women alike, occurring at a rate of about one in every 10,000 in most Western countries.
While science journalism is not, for the most part, intentionally fraudulent or misleading, it sometimes gives people the wrong impression about scientific findings by the way it interprets the data from recent articles in science journals like Cell and Human Molecular Genetics. When the Canadian scientists reported that they had inhibited an enzyme that cleaves, or cuts, the mutated HD protein (huntingtin) in mice, thus preventing the degeneration of the nerve cells in the brain, the press trumpeted it as a cure.
When the media takes such leaps or oversimplifies a complex, highly nuanced finding, it presents a skewed picture of the actual process of scientific research and discovery. In reality, the scientific method—that is, the process of empirical investigation into the validity or invalidity of a scientific claim or hypothesis—relies on replication and critical testing of each new finding, which takes a considerable amount of time. Not only does it require patience from both the scientists and the public, but it also requires a great deal of intensive effort that includes collaboration between research teams in different parts of the country and around the world.
Scientists rarely work alone or in isolation. To do so would be highly inefficient, especially since one scientist or group of scientists does not have expertise in every skill necessary to carry out an entire large experiment from start to finish. When teams of scientists work together and share ideas and materials (such as cell lines, which the MacDonald lab frequently sends to other labs), they are able to produce results in less time. However, “less time” does not mean instantaneously; collaborative work, while certainly more efficient than solitary work, still requires many years of sustained effort to find results that translate into good news for disease sufferers.
Although the scientific community values collaboration, it does not necessarily frown upon competition. Competition to test new ideas, to try and “knock them out of the ring,” is built into the scientific method (described later in The Scientific Method) and is, in a manner of speaking, one hallmark of the scientific endeavor.
Blueprints
One of the greatest rewards of scientific research is the “Eureka!” moment—that sudden gleeful breakthrough that can occur after much effort and many months of work. When a scientist experiences this lightning flash of insight, all the smaller discoveries of years past come together in a meaningful way, like the pieces of a puzzle, forming a much larger discovery. Indeed, HD can be compared to an enormous puzzle, the outlines of which are known, and the rest of which is still a mystery.
The genetic nature of the disease provides a kind of framework for discoveries about the changes that take place in the body on the cellular and molecular levels during the course of the disease. The macro-level changes observed in people with HD also provide guidelines about the micro-level changes occurring within their brains and bodies as the disease progresses.
With a rough outline to use as a guide, scientists can begin finding new pieces of the puzzle and fitting the puzzle pieces together to form recognizable pieces of the bigger picture. By putting the newly discovered pieces in place, scientists can make strides toward finding effective treatments not just for the symptoms of HD but also for the root genetic defect, or mutation, that causes the disease.
In 1993, an international team of researchers, which included Dr. MacDonald and her colleague, Dr. James Gusella, identified the responsible mutation. They found a CAG triplet repeat expansion in a region of human chromosome 4. Found in the nucleus (the information center) of cells, chromosome 4 is, like other chromosomes (we have 23 pairs of them), comprised of the DNA and associated proteins. Lengths of DNA in a chromosome make up genes, which are the functional units of heredity in humans and other organisms.
Each person inherits two copies (called alleles) of each gene, one from mother and one from father (the only exception being genes on sex chromosomes). Because HD is inherited as a genetically dominant character, a person needs only one mutated copy of the gene, called the expanded HD CAG allele, to inherit the disease. (For more information on genes and chromosomes, please click here
Genes are often compared to blueprints for making proteins. If the blueprint is defective, a defective protein will be made. Unlike the non-HD allele, which makes huntingtin protein with fewer than about 37 glutamines (one of the building blocks of the protein), the expanded HD allele makes an abnormal version of huntingtin, with an excess of glutamines (more than 37 or so of them in a row). Due to this mutation, the expanded glutamine version of the huntingtin protein does something—or is a byproduct of another process that does something—that contributes to the slow destruction of nerve cells in the brain. While the onset of symptoms can vary widely, the onset typically occurs between the ages of 30 and 50, after a substantial percentage of the nerve cells have died. There is also a juvenile form of the disease whose symptoms commonly appear before the age of 20. For more information on juvenile HD, please click here.
Physicians typically group the symptoms into three categories: movement, cognitive, and psychiatric. Movement symptoms include uncontrollable movements such as twisting and turning (known as “chorea”), rigidity, falling down, difficulty physically producing speech, and, in the later stages of the disease, difficulty swallowing, which can lead to significant weight loss. Cognitive symptoms include the altered organization and generally slowed processing of information in the brain. The most common psychiatric symptom of HD is depression; other symptoms include personality changes, anxiety, obsession, delirium, and mania. Denial of having HD is also a common symptom of the disease.
Presymptomatic genetic testing is available for those at risk for HD (i.e. people whose mother and/or father were diagnosed with the disease). While there is currently no cure for HD, there are drugs available to treat some of the symptoms, particularly chorea and depression. Some HD researchers, however, are beginning to develop and test drugs that target the presymptomatic effects of the genetic mutation that causes the disease.
Dr. MacDonald is one such researcher who works at the beginning of the disease pathway. She and Dr. Gusella, now director of the Center for Human Genetic Research, believe that the most effective treatments will be those that are specifically designed to reverse the first effects of the genetic mutation. These effects may impart altered physiology that is intrinsic to being born with and living with the HD mutation from birth. Scientists are still a long way from fully understanding the biology of the disease and the underlying mechanisms of nerve cell degeneration.
The Scientific Method
HD research can also be compared to erecting a building without knowing its dimensions. As of now, researchers only have a vague idea of the shape of the “building,” as specified by the genetic information on chromosome 4. In time, as scientists learn more about the cellular and molecular basis of the disease, they will have a clearer idea of what the “building” actually looks like.
Creating a firm foundation, as the first order of business, is key. Before moving forward with his or her research, a scientist must look to the relevant data from past research and attempt to replicate the key results of other scientists. This step is to make sure that the foundation is sturdy before beginning to build the first wall, or the second. And after completing the second wall, one must make sure that it does not fall when the wind blows, so to speak, in the face of different experiments designed to knock it down.
The idea is to avoid building a flimsy house of cards, but rather to make a solid structure that can be inhabited (and tested) for many years and decades by future generations of scientists. Progress can be thought of either as building a new piece of the foundation that may not initially be connected to the rest, or it can be the addition of new pieces to the growing structure on the original foundation. Progress is accomplished by employing the scientific method as follows:
- Repeat earlier findings in your specific area of inquiry.
- Make one or more hypotheses—that is, succinct propositions about what you expect to find if you are right about a process or phenomenon. Be sure your propositions are suitable for empirical testing in laboratory experiments.
- Conduct an appropriate experiment, controlling for (that is, holding constant) conditions other than those specified in your hypotheses. Carefully observe and note what happens in detail. These details, in combination with the conditions under which they were obtained, are your results.
- Check your results against the original hypotheses: Do they support one or another of your propositions? Are they what you expected or predicted from one argument or another? Or do they require that you reject all your original hypotheses because you saw something new or unexpected?
- Explain what you saw and what that tells us. You may need to modify the original hypotheses or, if necessary, you may need to make entirely new ones that are consistent with your findings.
- Repeat this process until you have eliminated all but one remaining hypothesis. Note that the last hypothesis “left standing” is not what we might call “proven”; it is simply our best bet, given current knowledge. It, too, may be rejected one day when we have better information and understanding.
Although not all research progresses in such a linear fashion, the scientific method can nevertheless be conceptualized as a flowchart:

Fig. 1. “How scientific investigations proceed.” (from Jones et al, 1994.)
The most time-consuming aspect of research—indeed, the heart of any scientific endeavor—is the continual knocking down and building up of the various parts of the knowledge-structure. Scientists can only make progress by first attempting to disprove previous hypotheses, including their own, to ensure the strength of the structure’s foundation. Researchers must also allow time to investigate unexpected results and decide how they fit (or don’t fit) into the emergent structure.
Overall, the scientific method provides scientists with an orderly, systematic way of approaching their research that, in the end, guarantees progress. But it is a multi-step process that cannot be shortened as a result of pressures either from the scientific community or the public without weakening the entire structure. The pace of research can, however, be accelerated by adding more trained scientists, by building and using machines that can allow experiments or observations more quickly and without bias, and by increasing the rate of flow of accurate information about the research, both within the scientific community and from scientists to the public which, directly or indirectly, funds most of these efforts.
Genetic Research at Massachusetts General Hospital (MGH)
The MacDonald lab is located in the brand-new Richard B. Simches Research Center, just up the street from the main MGH campus. Designed to facilitate communication between the various research groups, the building features wide hallways, open spaces, and meeting rooms equipped with audiovisual equipment for presentations. A spiral staircase, representing the double helix structure of the DNA molecule, connects the fifth and sixth floors, which make up the Center for Human Genetic Research (CHGR), through to the seventh floor, which houses the Molecular Biology Department.
One of five new thematic centers launched at the Hospital, the CHGR strives to facilitate the genetic research cycle, which begins with basic research, driven by scientists’ interest in questions pertaining to the biology behind a genetic disease. In basic research, biologists try to make new discoveries about the disease. For example, by studying animal models relevant to a given disease, scientists can try to observe new phenotypes in animals (that is, observable properties, particularly those associated with gene effects) that can then be looked for in human patients as well. Or researchers may try to use these new phenotypes to develop novel assays (chemical analyses) that can be used to discover drug compounds that may prevent the disease-associated phenotype.
The next stage of the cycle is the applied, or engineering-type, research, which puts the discoveries of basic research into practice. For example, researchers, usually in biotechnology or pharmaceutical companies, may use a variant of the assay discovered in the academic research lab to test a wide variety of drug compounds to see which of them effectively alter the outcome. Then, they may give the effective ones to animals and evaluate the outcomes, modifying the compounds by changing the chemical structure and retesting them, in successive rounds, to make them perform better, with fewer untoward side effects.
At this stage, researchers often look for drug targets, or molecules that can be expected to enhance or inhibit the disease. The best drug targets provide a direct route to what should be changed in a patient on the molecular level. Testing drugs in animal models helps researchers to identify targets and prioritize the best ones for further testing.
The third stage of the research cycle is clinical research, in which physicians and clinical researchers administer drugs to patients in government-approved clinical trials. Observations made at this stage often give rise to hypotheses at the basic research stage, and the cycle begins again, as illustrated in the diagram below.

Fig. 2. The genetic research cycle.
The cycle begins with basic research in academic labs, continues with applied research in biotech or pharmaceutical labs, and ends with clinical research in hospitals. Observations from the clinical phase may be used in basic research, and the cycle begins again.
Genetic research, or research of any kind, is therefore not monolithic; there are various stages of the research effort that operate in different facilities, with different kinds of people, and on different timelines for completing experiments and trials. Because each type of research has different goals, it requires funding from different sources.
The HD researchers at the CHGR are among the many people at MGH working to facilitate the genetic research cycle for HD. The MassGeneral Institute for Neurodegenerative Disease (MIND), directed by Dr. Anne Young, Chief of Neurology, makes discoveries in the basic realm and aims to translate them into prevention and treatments of neurodegenerative diseases like HD and Alzheimer’s. MIND, therefore, serves as a bridge between basic and clinical research. On the clinical side, the Department of Neurology helps HD patients to manage their symptoms through medical treatment, such as drug regimens, some of which may be experimental, in the cadre of clinical trials.
The study of HD at Massachusetts General Hospital via the scientific method can be compared to what scientists call a “fractal,” a geometric pattern that is repeated at ever-smaller scales, as in the diagram below. Whatever the size or scale of the problem—whether a researcher is looking at a molecule, an organism, or an entire population—the process has a regular structure (derived from the scientific method) and resembles the greater whole.

Fig. 3. “Construction of a Fractal Snowflake.” (from MSN Encarta.)
The basic triangle shape is reflected at every stage in the process of forming the larger design, just as the scientific method is reflected at every level of research from the smallest to the biggest detail.
As part of the CHGR, the MacDonald lab performs basic research and takes a molecular genetic approach to understanding HD. The researchers examine the DNA sequences of genes—the HD gene, in particular—to understand how changes in gene expression and protein structure are affected by the HD mutation. Gene expression is the process by which a gene’s DNA sequence is converted into proteins that are involved in cellular processes both structurally and functionally.
Studying the genetic expression of the HD gene (both the HD and non-HD causing alleles) can provide scientists with clues about how the nerve cells stay healthy or get sick. Determining the temporal order of the early steps in the disease pathway will eventually lead to the development of drug compounds that prevent these steps from occurring. As biological models for the disease, the MacDonald lab uses genetically altered mice and cells derived from them. Because the mice have high numbers of glutamine repeats in the huntingtin protein, as a result of the same HD CAG mutations that cause HD, they are likely to reveal the earliest presymptomatic changes to manifest with HD in humans.
From Journalism to Science
When I arrived at the lab at the beginning of July, I was eager to make a discovery of my own that would, in some small way, help scientists to find a cure for HD. I imagined myself working feverishly under the fume hood, swirling neon-colored chemicals in Erlenmeyer flasks. I envisioned myself peering into a high-powered microscope to observe the elusive structure of the huntingtin protein. As ridiculous as it sounds, I even imagined jumping up from my chair and crying, “Eureka!” as I bounded down the hallway in triumph.
As I settled into the daily routines of the lab, however, I saw the fantasy evaporate before my eyes. My biggest discovery this summer was that, while some discoveries may come in the form of intense flashes of insight, this is a rare event—except in the movies, of course. Getting to this point is much more prosaic.
Working in a lab was a brand-new experience for me. I am, however, familiar with the biology behind HD. For the past three years I have worked for Huntington’s Outreach Project for Education, at Stanford (HOPES), a student-led educational service project working to build a global Web resource on HD. Our site is a “layperson’s guide” to the scientific intricacies of HD and HD-related research. Akin to science journalism, my work has consisted of writing news briefs on the latest research, drugs, and other treatments, as well as interviewing eminent scientists and writing articles about their work on HD. My first interview was with Drs. MacDonald and Gusella during the summer of 2004, published on the website as the first chapter in a section called Research Frontiers. The two researchers discussed at length their approaches to HD and touched upon some of the myths of scientific research, including the ever-popular notion of a sudden cure or “magic bullet.”
Several months ago, just prior to my graduation from Stanford, Dr. MacDonald invited me to do an eight-week internship at her lab in this summer. After earning my BA in English with a minor in Human Biology, I headed east to Boston to begin my work. I was going from writing about science to actually doing science—a big leap.
Dr. MacDonald introduced me to Drs. Gill Gregory and Surya Reis, the two postdoctoral fellows who would be supervising my independent project. In preparation for conducting future research in their area of specialty, postdocs are in the last phase of their training, preparing them to start their own research laboratories, each working on a piece of the research puzzle. Research technologists, on the other hand, are responsible for performing one or more experiments that may either be varied or more routine, requiring long-term concerted expertise. For instance, Lakshmi Mysore, who has been working on HD for twenty years, specializes in genotyping, or determining the genetic makeup of an organism. The lab also depends upon the work of animal technicians, such as Edith Toral Lopez, who oversees the breeding of the animals and provides the genetically altered mice used in experiments.
During my first week I was outfitted with a white lab coat and notebook, given a tour of the lab, briefed on safety procedures and experimental protocols, and taught basic lab skills such as pipetting (using a syringe-like instrument to measure and transfer liquids from one container to another), taking care of tissue cultures, and transferring cells onto cover slips to be mounted on slides for viewing under the microscope.
Tissue cultures are a means of keeping populations of cells alive outside the body in a nutrient-rich liquid called a medium. I was responsible for monitoring the cells’ growth rate from day to day and splitting up the cells onto new dishes with fresh medium when the old dishes became too full because the cells had multiplied. The cells came from the brains—specifically, the striatum, the part of the brain that is first affected in HD—of mutant mouse embryos (those with 109 glutamine repeats in the huntingtin protein) and their normal (“wild-type”) counterparts.
My Summer Project
My task as an intern would be to complete a small project within the context of Surya’s and Gill’s research. Each of the postdocs takes a slightly different approach to detecting the subtle differences between mutant and wild-type nerve cells. Surya uses immunocytochemistry (IC), a method of staining cells with antibodies so that she can pinpoint the location of the huntingtin protein, for example, in the nuclei. Meanwhile, Gill uses immunohistochemistry (IH), a method of staining tissue slices (from the striatum, in this case) with the same antibodies, also to locate huntingtin in the nerve cells.
Antibodies are proteins made by the body’s immune system as a defense against foreign material, such as bacteria or viruses, which enters the body. These Y-shaped proteins attack and neutralize the substances, called antigens, that triggered the immune response. Each antibody has a specific antigen to which it binds. The IC and IH methods make use of an antibody’s ability to recognize a particular antigen, rather than its ability to attack and neutralize it. Please see below for a diagram of an antibody.

Fig. 4. The structure of an antibody. (from Wikipedia.)
To visualize the location of the huntingtin protein in the nucleus of a mouse nerve cell, researchers use a technique called immunostaining as part of the IC and IH methods. After fixating, or preserving, a cell sample or tissue slice on a cover slip or slide, they add a small amount of a primary antibody. The primary antibody recognizes and binds to a specific place on the huntingtin protein’s surface, called an epitope. Then, a secondary antibody that comes from another animal is used to detect the first. The secondary antibody contains a fluorescent molecule, which allows the researchers to see the position of the huntingtin in the cell under the powerful confocal microscope. Multiple secondary antibodies bind to the primary, thereby amplifying the fluorescent signal. Please see below for a schematic diagram of immunostaining.

Fig. 5. Immunostaining.
The primary antibody recognizes the polyglutamine tract of the huntingtin protein, and the secondary antibody recognizes the primary. The secondary antibody contains a fluorescent molecule to help scientists visualize the huntingtin under a microscope. Multiple secondary antibodies bind to the primary for fluorescent signal amplification.
Using confocal microscopy, scientists can visualize the huntingtin protein molecules to which the primary antibody binds. They can therefore show the localization of huntingtin and make conclusions as to the site-specific function of the protein (both mutant and wild-type) in the cells.
My project fit neatly into this experimental framework and had two objectives. The wet lab component of the project, performed at the lab bench, involved immunostaining with a primary antibody made at two different commercial laboratories. My goal was to determine which of the two versions was better, and under what conditions, for seeing differences between the wild-type and mutant cells with regard to the staining pattern. See below for a picture of me performing an immunostaining procedure.

Fig. 6. Taylor Altman performs an immunostaining procedure.
She applies a primary antibody stain to mutant and wild-type cells on cover slips.
The dry lab component, performed at my desk, entailed building an electronic database of information about huntingtin antibodies. In a Microsoft Excel spreadsheet, I organized the information by such categories as antibody name, epitope, and animal host (the animal from which the antibody is taken). The database will eventually be turned into a website for use by HD researchers all over the world.
Accomplishments
Before long, I realized that my goals were unrealistic for the two months I’d be spending at the lab. I saw that I couldn’t build an entire database on huntingtin antibodies in one summer because there are hundreds that have not yet been properly described, and information is often scarce. I did manage to gather sufficient data for eight antibodies, which is a good start.
As for the wet lab project, I completed three modest experiments, each with its own purpose and goal, on the path to determining which of the batches of primary antibodies was better suited to seeing differences between the mutant and wild-type striatal cells. Although I was a bit disappointed that I couldn’t see my project out to its end, I did get a good feel for bench work and for the extensive planning that goes into each experiment.
The first of my small experiments was a primary antibody dilutions test. Dilution is the process of making something weaker or less concentrated. To get different dilutions of the antibody, I added the same amount of antibody to increasingly large amounts of the diluting solution. Then, I applied the dilutions to the mutant and wild-type cells to determine the optimal dilution for seeing the greatest differences between the two types of cells under the confocal microscope. As the dilutions increased, I expected the strength of staining to decrease more rapidly in the wild-type than in the mutant. However, I saw the exact opposite. See below for pictures of the wild-type and mutant cells at the highest and lowest dilutions. Notice the difference in strength of staining between the two.

Fig. 7. Wild-type and mutant cells at lowest and highest dilutions.
Pictures taken at 20x magnification on the confocal microscope. As the dilutions increased, the strength of staining decreased more rapidly for the mutant than the wild-type.
Part of science is dealing with unexpected results. When a scientist initially gets results that seem to go against the hypothesis, he or she must repeat the experiment in order to rule out chance or human error. My second experiment, therefore, was a repeat of the first. Again, I got the same results. Clearly, this was no coincidence.
To look for clues that might explain my results, Surya directed my attention to the method I used to fixate the cells. Using a detergent, I had made tiny holes in the cells’ membranes that allowed the primary and secondary antibodies to flow in. Perhaps I’d used too much detergent and had made the membranes too porous, thereby letting in too much antibody or letting out too many important cellular components. Or there may have been other explanations for the results—maybe the cell types had been mixed up, or maybe the antibody no longer worked. The latter seemed most probable.
My third and final experiment, then, was a test of the amount of detergent, in which I kept the antibody dilutions the same while I varied the concentrations of detergent. On the whole, the results were inconclusive, but Surya plans to run another test in the near future.
Conclusions
Reflecting on my summer at the lab, I am grateful to Gill and Surya, and to their supervisors Marcy McDonald and James Gusella, for the rewarding experience I had at MGH. Not only was I able to learn about the process of scientific discovery firsthand, but I was also able to become a better science writer by enhancing my knowledge of scientific materials, methods, and terminology. By conducting my own experiments, I began to think like a researcher and to understand the scientific method from the point of view of one who puts it into practice on a daily basis.
I saw a side of science that the public rarely, if ever, sees. The actual process of scientific discovery is much lengthier, more complex, and more nuanced than the media’s portrayal of it. Science is about careful observation and planning, good record keeping, and building a strong foundation for future experiments by continually attempting to disprove previous hypotheses.
Above all, science requires great patience and perseverance. Scientists do not leap from their lab benches crying, “Eureka!” every day, nor do they produce a continual stream of results. Moreover, results do not come cheaply. To my surprise, I learned that it cost about $5,000 for the supplies and small equipment, and about $400,000 for the confocal microscope, to perform one of my “simple” immunostaining experiments (not to mention the still-higher cost of paying everyone’s salaries and keeping the lab running on a daily basis).
Although my future plans don’t include working as a researcher, science will always be a part of my life, whether I choose to become a science journalist or simply a science enthusiast. I now have a much greater understanding and appreciation of the arduous work that scientists do, which gives me hope that someday they will solve the mystery of HD and other neurodegenerative disorders.
For further reading
- “Construction of a Fractal Snowflake.” MSN Encarta. 28 Aug. 2006
<http://images.encarta.msn.com/xrefmedia/aencmed/targets/illus/ilt/T046583A.gif>.
- Jones, Allan, Rob Reed, and Jonathan Weyers. “How scientific investigations proceed.”
Practical Skills in Biology. Essex: Addison Wesley Longman Limited, 1994.49.
- “Schematic of antibody binding to antigen.” Wikipedia. 21 Aug. 2006
<http://en.wikipedia.org/wiki/Antibody>.
- Ubelacker, Sheryl. “Canadians cure Huntington’s disease modified mice.” The Globeand Mail. 16 June 2006. 15 Aug. 2006
<http://www.theglobeandmail.com/servlet/story/RTGAM.20060616.whuntingtonS016/BNStory/Science/home>.
T. Altman, 2006
More
Introduction
This chapter will investigate how cholesterol relates to HD. The chapter begins with a general overview of cholesterol and its role in the body. Following this, the chapter will focus on the cholesterol that originates in the brain, and on new research that looks at the relationship between cholesterol in the brain and HD.
What is cholesterol and what does it do?
Cholesterol is a lipid molecule present in all animals. It is largely found in cell membranes, and there is a smaller amount circulating in the blood stream and stored inside cells. Cholesterol has a number of important functions. It is a key structural component of cell membranes, maintaining their fluidity and stability, and enabling important processes such as endocytosis. It is also important for the metabolism of fat-soluble vitamins, the manufacture of bile salts and the synthesis of vitamin D and steroid hormones. The synthesis of vitamins and hormones takes place in endocrine cells, while bile salts are generated in the liver.
Recently a small number of papers have shown that HD patients have altered levels of cholesterol in nerve cells. Since cholesterol plays a key role in the maintenance of healthy neurons, the disruption of normal cholesterol levels in HD patients may be a significant cause of neuron death and dysfunction.
Where does cholesterol come from?
There are two major ways for our bodies to get cholesterol; it can be synthesized in the body, or obtained from the diet. Normally, our bodies take advantage of both methods of getting cholesterol. On average, a 150 pound person will synthesize about 1 gram of cholesterol per day and intake 200-300 milligrams through their diet.
The highest rate of cholesterol synthesis by the body occurs in the liver, although cholesterol is also made in the intestines, adrenal glands, CNS, and reproductive organs. Other cells can produce cholesterol, but typically in much lower amounts.
Cholesterol is found in all animal foods including meat, poultry, fish, seafood, eggs, and dairy. Cholesterol is not found in plants, so foods like fruits, vegetables, grains, nuts and seeds do not raise cholesterol levels. It is partly because we synthesize so much of our own cholesterol that excess dietary cholesterol is not necessary and can be harmful in a variety of ways.
In this chapter, our goal is to first provide a general review on cholesterol and its activity in the human body, and then look at its relationship to Huntington’s disease.

HDL and LDL
Most people have heard of a distinction between two types of cholesterol: high-density lipoprotein (HDL) cholesterol, and low-density lipoprotein (LDL) cholesterol. HDL is commonly referred to as “good” cholesterol, while LDL is called “bad” cholesterol. More precisely, HDL and LDL are not simply different types of cholesterol, but rather alternative groups of lipids and proteins that transport the cholesterol throughout the body in the bloodstream. Molecules such as HDL and LDL are needed to carry cholesterol because it is a hydrophobic molecule and therefore cannot dissolve in blood and travel through the bloodstream on its own.
But if HDL and LDL are just alternative cholesterol carrier molecules, why is one considered good and the other bad? Medical studies have noted that high levels of LDL are associated with an increased risk of cardiovascular disease, whereas high levels of HDL are associated with decreased risk of cardiovascular disease.
How exactly does HDL produce beneficial effects and LDL produce harmful effects? LDL is the major cholesterol carrier in the blood and is responsible for delivering cholesterol to cells in the body. High levels of LDL cholesterol in the blood contribute to the formation of plaque. Plaque is a thick, hard deposit of fat, cholesterol and other substances that clogs arteries and causes atherosclerosis. If arteries become severely clogged with plaque, oxygen-carrying blood may not reach be able circulate around the body- which can lead to heart attack or stroke. Approximately one fourth of blood cholesterol is carried by HDL. HDL is believed to protect against atherosclerosis by carrying cholesterol away from the blood (so it cannot contribute to plaque formation) or even removing excess cholesterol from plaque already built-up in the arteries. HDL usually delivers cholesterol to the liver or endocrine cells, where it will be used in the synthesis of steroids or bile salts, and ultimately removed from the tissue and bloodstream.
Cholesterol as a Risk Factor for Heart Disease
When our cholesterol levels are tested, they are shown in milligrams per deciliter of blood (mg/dL). The American Heart Association classifies anyone with total cholesterol greater than or equal to 240 mg/dL as belonging to a high risk category. They recommend that those with a total cholesterol level in this high range get a complete fasting lipoprotein profile done. This test measures LDL, HDL, and triglyceride levels. Triglycerides are another contributor to atherosclerosis. The target HDL level is greater than 40 mg/dL, the target triglyceride level is less than 150 mg/dL, and the target total cholesterol level is less than 240 mg/dL.
Cholesterol and Triglyceride Levels (mg/dL)
|
Optimal |
Near Optimal |
Borderline High |
High |
Very High |
| Total Blood Cholesterol |
<200 |
—- |
200-239 |
=240 |
—- |
| LDL Cholesterol |
<100 |
100-129 |
130-159 |
160-189 |
=190 |
| Triglyceride Level |
<150 |
—- |
150-199 |
200-499 |
=500 |
*Information from the American Heart Association
There are several ways to lower cholesterol levels that are too high. The best methods are usually lifestyle changes. These can include dietary changes such as eliminating foods that are high in saturated fat, trans fat, and cholesterol and increasing the consumption of fruits, vegetables and grains. Exercise is also an important way to reducing the amount of cholesterol in our bodies. By exercising for 20-30 minutes each day we use up greater amounts of fats and other energy molecules that are stored in our bodies. Additionally, there are medications that help lower cholesterol. These medications usually employ one of two general strategies. They either block the synthesis of cholesterol within the bodies’ cells or they prevent cholesterol uptake in the intestine, forcing ingested cholesterol to pass through the body and never be absorbed. The best way to stay healthy is to make sure you have had your cholesterol tested and, if it is too high, to follow your doctor’s instructions for lowering it.
Cholesterol in the CNS
The CNS contains a large amount of cholesterol, as cholesterol is needed for the growth and maintenance of myelin, as well as neuron and glial cell membranes and for the formation of new connections between cells. However, the CNS is unique in that there is no evidence that it obtains any of its cholesterol from the blood. Instead, cells in the CNS synthesize all of their own cholesterol. In fact, the rate of cholesterol synthesis in the CNS exceeds the need for new cholesterol, so that some cholesterol must move out of the CNS through excretory pathways.
It is not easy for molecules to enter the CNS. Tightly joined endothelial cells found in the capillary network within the brain prevent many molecules from moving from the blood to the CNS. This blood-brain barrier makes it unlikely that cholesterol carried in lipoproteins could reach the CNS unless there were specific transporters in the endothelial cells of the vessel walls. Currently there is no evidence that existing transporters in those endothelial cells actively uptake lipoprotein-transported cholesterol.
Relating cholesterol to Huntington’s disease
A few studies have recently investigated the role of cholesterol in HD and have suggested that HD may disrupt the normal cholesterol homeostasis in the brain. These research articles propose that the altered huntingtin protein may cause a change in intracellular levels of cholesterol in neurons by disrupting at least two cellular mechanisms: endocytosis and cholesterol biosynthesis. Ultimately, these cellular changes may lead to dysfunction or death of the striatal neurons and reflect another pathway or mechanism by which the mutated huntingtin protein affects the cell and causes neurodegeneration.
Cholesterol Accumulation and Inhibited Endocytosis
A study by Trushina et al. has reported that the mutant huntingtin protein inhibits a specific type of endocytosis in striatal neurons. These neurons are also shown to have strikingly high intracellular levels of cholesterol.
Mutant huntingtin has been previously shown to interact with clathrin, which is a major protein involved in endocytosis. In this study however, a different protein has been implicated in the disruption of endocytosis in HD. It has been demonstrated that the mutant huntingtin protein interacts with the protein caveolin-1 (cav1), a key molecule in a different endocytotic pathway (called caveolar-related endocytosis). The interaction of mutant huntingtin protein and cav1 inhibits caveolar-related endocytosis and also causes an accumulation of cholesterol within neurons.
Examination of mouse tissue and HD striatal cell cultures revealed the accumulation of intracellular cholesterol. Researchers found that using siRNA to knockdown cav1 translation prevents cholesterol accumulation. For more on siRNA techniques, click here. This occurred only in the continued presence of mutant huntingtin protein, suggesting that it is something specifically about the nature of the interaction between altered huntingtin and cav1 that disrupts normal cholesterol homeostasis, and not simply the lack of cav1 altogether. It was also observed that in all cases clathrin-dependent endocytosis was normal, indicating that the mechanism of cholesterol accumulation was specific to the disruption of the caveolar-related pathway.
How is cholesterol biosynthesis affected?
In another recent paper, by Valenza et al., Huntington’s disease has been shown to decrease cholesterol biosynthesis in nerve cells. The presence of altered huntingtin in these cells is correlated with significantly lower total cholesterol mass. This was observed in mouse tissue and in cultured striatal neurons expressing a fragment of the mutant huntingtin protein.
Mutant huntingtin affects the transcription of genes crucial to cholesterol synthesis. The altered huntingtin protein interacts with binding proteins called sterol regulatory element -binding proteins (SREBPs) and prevents these proteins from entering the nucleus. These proteins usually bind to DNA and promote transcription of many different genes important for synthesizing cholesterol. Mutant huntingtin has a strong effect on SREBPs; the proteins are reduced by 50% in the nucleus of HD cells. Reduction of the SREBPs results in significantly less transcription of the genes involved in cholesterol biosynthesis, which ultimately reduces total cholesterol.
Large changes in the levels of intracellular cholesterol will eventually lead to disruption of cellular homeostasis. Research with HD cell line models has shown that the addition of exogenous cholesterol to cultured striatal neurons expressing mutant huntingtin joined to a green fluorescent protein will prevent these neurons from dying.
Implications
Cholesterol is essential for promoting synapse formation and maintaining membrane integrity in CNS neurons. It is also a major component of myelin and important for optimal neurotransmitter release. Because cholesterol plays such a major role in CNS growth, development, and maintenance, disruptions of cholesterol homeostasis can have negative consequences. Accumulation and depletion of intracellular cholesterol in neurons are both possible mechanisms contributing to neuron dysfunction in these HD models. However, the findings are limited to HD cell models and postmortem HD tissue. This work now needs to be followed up by investigating these changes in HD patients to see whether similar dysfunction occurs.
If studies in human subjects found a similar dysfunction in cholesterol homeostasis, it might suggest that adjusting the cholesterol levels in neuronal cells could be investigated as a potential treatment for HD. Future research may aim to discover how to transport cholesterol across the blood-brain barrier and whether cholesterol therapy could be one way of slowing or halting neuronal cell death in HD.
It is interesting to note that similar defects in caveolar-related endocytotic pathways and perturbations of cholesterol homeostasis have been implicated in other neurodegenerative diseases related to HD like Alzheimer’s disease and Parkinson’s disease.
Summary
Recent research has suggested that disruptions in cholesterol homeostasis could be important in explaining how the HD mutation causes neurodegeneration. However, cholesterol’s role in the disease is still not fully understood. It might seem strange that HD has been linked to both intracellular cholesterol accumulation and depletion. One current hypothesis is that different stages of the disease are characterized by different disruptions to cholesterol homeostasis. Future research should shed light on the connections between these different disruptions and normal cholesterol activity.
For Further Reading
-A. Hepworth, 5/13/2007
More
Dr. Elena Cattaneo
Department of Pharmacological Sciences and Center of Excellence on Neurodegenerative Diseases, University of Milan
Milan, Italy
In September of 2004, HOPES team members Devon McGee and Agnieszka Milczarek visited the Laboratory of Stem Cell Biology and Pharmacology of Neurodegenerative Diseases, headed by Dr. Elena Cattaneo, at the University of Milan in Italy. HOPES would like to thank Dr. Cattaneo and the rest of the members of the lab for taking time out of their busy schedules to meet with us and share with us the philosophy behind their research. The following is the lab´s mission statement:
The ingenuity of mankind, often anonymous, has taught humanity to always make better use of water which runs ceaselessly… The mill is the processor of the river directing simplicity and chaos into a powerful and productive force. Our laboratory strives to remain a mill in constant operation.
Introduction
Dr. Elena Cattaneo did her graduate work at the Massachusetts Institute of Technology in the lab of Professor R.D.G. McKay, where she became interested in neural stem cells and their potential therapeutic applications to neurodegenerative diseases. She decided to focus her studies on neural stem cells from a human brain structure called the striatum because she realized that “the striatum is at the center of many biophysiological events.” The striatum forms part of the basal ganglia, and is the part of the brain most affected in HD. (For more information on the basal ganglia and their role in HD, click here.) Remarkably, while at MIT, she was able to “immortalize” stem cells from the striatum and create a new cell line called ST14A. The cells exhibit quite accurately many of the properties of nerve cells from the striatum and can be engineered to express either normal or mutant huntingtin. (For more information about huntingtin, click here.) The cell line offers a great model of the Huntington´s disease and researchers around the world now use it in their labs.
Presently, Dr. Cattaneo works in the Department of Pharmacological Sciences and in the Center of Excellence on Neurodegenerative Diseases at the University of Milan, where she is a full professor of pharmaceutical biotechnology. Since 1995, she has collaborated on Coalition for the Cure, an international research group organized by the Huntington´s Disease Society of America. (For more information on the Huntington´s Disease Society of America, click here.) She has also collaborated on the Cure HD Initiative sponsored by the Hereditary Disease Foundation. Since 1988, she has been an investigator in the Italian Telethon Foundation. Dr. Cattaneo has received many awards and distinctions, including the “Le Scienze” Prize for Medicine and a medal from the president of Italy for her work on stem cells and HD. In addition, she coordinates a national program on stem cells and participates in Eurostemcell (www.eurostemcell.org), a group for research on stem cells funded by the European Union.
Dr. Cattaneo´s lab is comprised of senior scientists (similar to post doctoral fellows), junior scientists (similar to graduate students), and undergraduate students. Overall, there are about fifteen people working in the lab. Each member of the lab chooses and proposes his/her own research project to work on. Although the members of the lab have diverse research interests, the lab is unified in its quest to someday find a cure for HD. There are weekly lab meetings and each person is at least partially involved in all ongoing research projects.
Currently, the lab is focused on the mechanisms of neurodegeneration in Huntington´s disease, as well as neural stem cell biology and its potential application to HD and other neurodegenerative diseases. (For more information on stem cells and their potential to treat HD, click here.) The ultimate goals of the lab are to identify cellular pathways that might be suitable for therapeutic intervention and to find new methods for drug testing. While a majority of HD research is focused on the harmful effects of the mutant huntingtin protein, Dr. Cattaneo´s lab looks at the other side of the problem by examining the normal function of huntingtin protein. This is important because not much is known about how the huntingtin protein is supposed to work normally. By investigating the normal function, the researchers hope to find new ways of treating HD. Dr. Cattaneo´s research has revealed that normal huntingtin possesses a variety of anti-apoptotic capabilities, which means that it helps keep cells alive.
Thus, on the basis of Dr. Cattaneo´s work, the damaging effects of HD should be attributed not only to the toxic function of mutant huntingtin, but also to the loss of beneficial function of normal huntingtin. With this in mind, the lab hopes to completely make clear the physiological role of normal huntingtin and its mode of action in order to develop new ways to combat the disease. For example, once more is known about normal huntingtin, researchers can attempt to restore normal huntingtin function in people with HD, and this restoration of normal huntingtin function may significantly alleviate many HD symptoms.
Members of the research lab are also involved in the study of the biology of neural stem cells derived from the developing human brain. Neural stem cells seem promising because they are the developmental source of nerve cells. The number one problem in neurodegenerative diseases like HD is the death of nerve cells, which are usually unable to regenerate themselves. Neural stem cell transplantation may help restore nerve cells and may even prevent nerve cells from dying in the first place.
Neural stem cell therapy for neurodegenerative diseases is still a distant goal and many obstacles remain before it can be used as a treatment. One obstacle, for example, is that it is difficult to properly integrate neural stem cells into a recipient brain. Presently, Dr. Cattaneo´s lab is trying to identify the signals that instruct neural stem cells to undergo division and differentiate into nerve cells. Understanding the mechanisms that regulate nerve cell survival and differentiation will make it easier to develop ways to integrate neural stem cells into the brain. In addition to studying nerve cell division and differentiation, the lab is also producing cell and animal models of brain diseases for studies of cellular mechanisms and of drug efficacy.
What´s special to Dr. Cattaneo about HD research?
HD research involves a great deal of obligation to the HD community and this obligation is what drives Dr. Cattaneo and her coworkers to produce promising results in the lab. The lab often receives not only phone calls but also visits from individuals and families affected by HD, who bring assortments of jams and cookies and cakes to show their gratitude for the researchers´ work. Dr. Cattaneo and the other members of the lab recognize that it is important for the patients and their families to know that someone is constantly working on uncovering the mysteries of HD. For this exact reason, Cattaneo and some of the other researchers occasionally present their scientific results, in non-technical terms at the meetings of Italian associations for people with HD. This constant interaction between the researchers and individuals affected by HD has such a positive effect on the lab that the researchers become not only “driven by curiosity, but also by the desire to help patients.” All the researchers in the lab confess that they feel increased pressure to produce and publish results because they know that they are working for real patients – and this “healthy pressure,” as Dr. Cattaneo calls it, is one of the factors that make HD research so special.
Dr. Cattaneo also believes that HD research is extraordinary because of the close-knit HD research community that continues to develop around the world. Funding and support from associations such as the Hereditary Disease Foundation and the Huntington´s Disease Society of America (HDSA) in the United States and others in the European Union help bring together researchers who share one goal – to find a cure for HD – so that they can work as a team rather than compete with each other for results. Since 1997, the HDSA has coordinated “Coalition for the Cure,” which organizes researchers from almost twenty different laboratories and organizes them into five teams that work on different aspects of HD. Cattaneo says that coalitions such as these not only promote but also require sharing of unpublished results among research labs, which helps the coalition as a whole make progress as researchers build on each other´s ideas. Furthermore, labs that are part of such alliances have the advantage of sharing research materials such as useful genes or experimental mouse strains (for more information on animal models in HD research, click here), which speeds up the research, keeps costs down, and makes possible experiments that might not otherwise be possible. This type of sharing helps research labs circumvent financial and legal obstacles that might prevent them from obtaining materials developed by other research institutions.
On the one hand, being part of a scientific community that emphasizes sharing goes a long way to reduce competitive barriers commonly seen between labs working on similar projects. On the other hand, Cattaneo says it also adds pressure to be constantly productive. Researchers are expected to present new results at each of the annual or biannual meetings they must attend for each coalition or organization. Cattaneo admits that such expectations can add stress to her life, but being part of a community of researchers who have placed complete trust in each other and have vowed to work together for families affected by HD makes it worthwhile. She emphasizes that the formation of such a close-knit community could only have been possible with a disease like HD, because there has been historically much less interest in HD research than in diseases such as Alzheimer´s or Parkinson´s. (For more information on these diseases and how they compare to HD, click here.) Through her work on HD, Dr. Cattaneo has forged strong relationships with researchers from across the globe, including Drs. James Gusella and Marcy MacDonald at Massachusetts General Hospital, whom two other HOPES members visited in the summer of 2004. (For more information about the latter, click here.) In the process, Cattaneo´s obligation to patients has been complemented by the obligation she now feels to produce valuable results that she can share with fellow researchers.
Approaching HD
According to Dr. Cattaneo, to be conceptualized correctly, HD must be seen as a cascade in which every single disease event leads to more events that cause further dysfunction and damage (see Figure AH-0.) In some way, HD researchers are lucky because they have a definite single starting point of the disease: a CAG expansion in the Huntington gene. Cattaneo agrees that the genetic nature of HD makes it easier to research than other neurodegenerative diseases, which can have varying genetic and environmental causes. However, the disease cascade has yet to be completely figured out, and Cattaneo stresses that it will be important not only to find a majority of the disease events, but also to arrange them in the correct temporal order; that is, to figure out when each dysfunction occurs, what causes it, and what other dysfunctions it causes itself. As is evident from Figure AH-0, this will not be an easy task.
Researchers in Dr. Cattaneo´s lab are already working on putting together the schematic cascade, but Cattaneo says that the work is difficult since “every result leads to more experimentation.” With every new mechanism she and the researchers discover, there is more they need to find out about its causes and effects. Cattaneo says that often, “your brain goes faster than your hands,” and while she or her colleagues may form hypotheses and ideas rapidly, the funding and time may not allow for immediate experimentation to follow up on those ideas. However, one researcher in the lab, Chiara Zuccato, was recently very successful in figuring out a very important aspect of the disease cascade. After learning that normal huntingtin protein increases the amounts of brain-derived neurotrophic factor (BDNF) in nerve cells, she set out to find the mechanism by which it does so. Zuccato found that normal huntingtin increases the production of BDNF by indirectly inhibiting a molecule called NRSE, which normally prevents the production of BDNF (this finding will be explained in more detail in a later section).
Because BDNF is a protective growth factor in the brain and is depleted in the nerve cells of people with HD, researchers in Dr. Cattaneo´s lab are now focusing their energies on the functions of normal huntingtin and what prevents it from performing these functions in people with HD. This approach is different from the classic research approach that has typically focused only on mutant huntingtin. However, mutant and normal huntingtin may be linked earlier on in the disease cascade and Cattaneo´s lab is now on a quest to figure out how mutant huntingtin´s toxicity could cause normal huntingtin´s loss of function in HD.
Unfortunately, BDNF production is only one of many cellular processes that are disturbed in HD, and Cattaneo says real progress will be made when researchers are finally able to link together all the dysfunctions in a cause-and-effect relationship. Transcriptional dysregulation is a major disease mechanism in HD that affects the production of proteins needed by nerve cells (one of these is BDNF, mentioned above). Mitochondrial dysfunction, which causes abnormalities in energy metabolism, is another major disease mechanism. (For more information on problems with energy metabolism in HD, click here.) As of now, researchers only understand bits and pieces of each of these major mechanisms, and Cattaneo believes that mapping out precisely what events lead to these dysfunctions will be crucial in understanding HD and developing a cure.
Because HD involves a complicated cascade of events, Dr. Cattaneo believes that an effective cure will actually involve a combination of treatments. She is very hopeful about the use of new techniques such as RNA interference, which attack the disease as close as possible to the beginning of the cascade by “silencing” the mutant Huntington gene. However, Cattaneo doesn´t think that such techniques will ever be able to fully block the production of the mutant huntingtin protein. (For more information on RNA interference, click here.) Cattaneo says that “a cure with only one strategy is not realistic; to battle HD you need a more global approach.” In the future, she believes that the most successful therapy, which could turn into a cure, will involve different types of treatments at different stages of the disease. According to Cattaneo, these treatments will ideally begin before the onset of symptoms and change accordingly as the disease progresses from the nerve cell dysfunction stage to the nerve cell death stage. Future treatment of HD may include:
- Stage I: Early treatment with RNA interference to prevent expression of mutant huntingtin; continues throughout the following stages.
- Stage II: Dysfunction of nerve cells combated by drugs that prevent toxic functions of mutant huntingtin and drugs that restore the functions of normal huntingtin.
- Stage III: Nerve cell death combated by cell replacement therapy (possibly with the use of stem cells) as well as protective strategies involving therapy with growth factors such as BDNF.
Dr. Cattaneo believes that for patients, “a cure is anything that gives them more time,” and she hopes that a treatment regimen like the one outlined above could postpone and decrease the symptoms of the disease to such an extent that it could in fact be considered an effective cure.
However, because potential drugs will be used to combat the disease at different time points in different individuals, the researchers will need biomarkers. Biomarkers are biological indicators that can be measured in patients to determine the severity of disease and to show whether drugs are effective in specific stages of the disease. Measuring the severity of symptoms is not sufficient because symptoms can vary so widely between patients and between stages of the disease. A possible biomarker that the Cattaneo lab is considering using is the level and activity of the A2A receptor, a receptor molecule that is expressed by nerve cells in the striatum and becomes more active in the presence of mutant huntingtin. Another possibility involves measuring levels of BDNF, which can also be used to track the progression and severity of the disease.
Dr. Cattaneo´s lab tries to simultaneously identify drug targets as they learn about new disease mechanisms. She stresses that successful drug development cannot happen without a deep understanding of the disease mechanism that the drug is meant to attack. Therefore, she believes that studies on compounds such as nutritional supplements and their effects in people with HD are not as promising at they may seem since most of the studies are conducted without an understanding of how the supplements exert their effects. An understanding of the disease mechanism is essential to picking drug targets – molecules that the drug will act on to alter some pathway – that will be both effective and safe. The drug must act early enough in the pathway to successfully stop the disease cascade, but it must also be specific enough so that it does not cause significant side effects.
Main Findings
Dr. Cattaneo´s lab has contributed to significant breakthroughs in HD research. Prior to the research done by the Cattaneo lab, it was assumed that since the HD allele gives rise to mutant huntingtin, mutant huntingtin must be toxic to nerve cells and it must be the only reason that nerve cells die. Indeed, numerous studies support this notion. However, Dr. Cattaneo´s lab showed that mutant huntingtin is not the only reason that nerve cells die. In fact, it may be only half the reason. Whereas earlier research had simply disregarded normal huntingtin´s role in the HD disease process, the Cattaneo lab found that normal huntingtin is actually crucial to nerve cell survival in the brain. They have found that individuals with the HD allele experience nerve cell death not only because their cells produce toxic mutant huntingtin, but also because the normal huntingtin they produce loses its normal functions.
Dr. Cattaneo and two other members of her lab, Dorotea Rigamonti and Chiara Zuccato, are leading experts in the field of normal huntingtin function. They have amassed a remarkable amount of evidence supporting the notion that normal huntingtin is, in fact, beneficial to nerve cells. They began their research in this area by first inserting extra copies of either normal huntingtin protein or mutant huntingtin protein into nerve cells grown in culture dishes in the lab. In 2000, they reported that nerve cells overproducing normal huntingtin can survive even when deprived of key growth factors or when subjected to other conditions that would normally cause them to die. Furthermore, they found that normal huntingtin appears to keep nerve cells alive by stopping the cascade of events that usually leads to apoptosis, or programmed cell death. They therefore concluded that normal huntingtin serves as a kind of protein “lifesaver” for nerve cells.
Other researchers, such as Scott Zeitlin of Columbia University in the USA, have confirmed these findings by creating knockout mice that express neither mutant huntingtin nor normal huntingtin. In such cases, the mice develop severe brain damage. In this instance, the brain damage can be explained not by mutant huntingtin (because it isn´t present), but by the absence of normal huntingtin (which also is not present). Interrupting huntingtin production at various points in the mice´s lives also leads to apoptosis. Remarkably, mice lacking normal huntingtin display very similar neurological symptoms to mice that express mutant huntingtin, suggesting that the absence of normal huntingtin and the presence of mutant huntingtin might be different sides of the same coin.
The discovery of the importance of normal huntingtin function was a significant contribution to the field of HD research. However, it does not explain why nerve cells in the striatum are targeted preferentially in HD. The huntingtin protein is produced in many cells throughout the body. Since normal huntingtin does not function correctly in people with HD, and since it is present in all types of cells, scientists were puzzled by the fact that only nerve cells in the striatum were seriously affected by the disease. Intrigued by this question, Zuccato and her colleagues undertook an examination of brain-derived neurotrophic factor (BDNF). As discussed above, BDNF is a growth factor that is known to be crucial to the development and survival of nerve cells in the striatum. It is usually produced in the cell bodies of nerve cells in the cortex and then travels to the striatum along fibers that connect the two brain regions. With this is mind, the researchers hypothesized that there might be a connection between BDNF and huntingtin. Amazingly, they found that normal huntingtin stimulates the production of BDNF in nerve cells grown in lab culture dishes. Specifically, huntingtin seems to indirectly activate the “on” switch, or promoter, of the gene that encodes BDNF. When this gene is turned on, it prompts nerve cells to make more BDNF. Normal huntingtin enables the gene to be turned on by inhibiting a molecule called NRSE, which normally turns the gene off. Contrarily, mutant huntingtin does not inhibit NRSE, which means that NRSE stays around and keeps the gene turned off so that no BDNF can be produced. Zuccato and her colleagues confirmed this link between huntingtin and BDNF by carrying out experiments involving genetically engineered mice. They found that mice overproducing normal huntingtin have elevated amounts of BDNF in their brains, whereas mice with mutant huntingtin have lower than normal levels of BDNF in their brains.
The lab´s studies are both enlightening and intriguing because the findings do not necessarily conform to previous hypotheses about the disease. They reveal the true complexity of the disorder. In addition to generating mutant huntingtin which is thought to interfere with several crucial cellular proteins and systems, HD also somehow deprives the brain of normal huntingtin – which would otherwise turn on the gene for the growth factor BDNF and protect nerve cells from apoptosis. As mentioned previously, Cattaneo´s lab believes that mutant huntingtin´s toxicity and normal huntingtin´s loss of function may be related. In fact, another group of researchers has recently shown in genetically engineered mice that mutant huntingtin can destroy normal huntingtin. It is also clear that normal huntingtin interacts with the brain in complex ways and there are many aspects of huntingtin that need to be further explored. As Dr. Cattaneo noted, “Much additional research must be completed before these findings can help patients, and we want to be clear that this is not a cure. But we are optimistic that our work will help guide the development of new therapies, such as drugs to replace or boost the activity of normal huntingtin, or to increase levels of another brain protein.”
By more clearly exposing the complexities of HD, Dr. Cattaneo´s lab has opened the door for the production of better treatments for the disease. Many of the drugs currently available only alleviate some of the symptoms of HD and can have serious side effects. Furthermore, the drugs often treat one symptom only to make another symptom worse. For example, doctors often prescribe sedatives to control involuntary movements, but these drugs also decrease levels of the neurotransmitter dopamine in the brain, worsening the patient´s depressive symptoms.
As a result of the findings of Dr. Cattaneo´s lab, several more innovative treatments for HD are currently being tested, such as replacing the damaged nerve cells with transplants of fetal tissue or injecting neurotrophic factors such as BDNF into the striatum. The use of fetal tissues is very controversial and raises many ethical questions, but the preliminary results do look promising. Researchers at the School of Medicine in Creteil, France have transplanted fetal nerve cells into the striata of five people with HD. Three of the people improved significantly in terms of motor and intellectual function. Currently, clinical trials with a larger number of patients are being conducted.
Obstacles and Challenges
Although Dr. Cattaneo certainly enjoys the rewards of being a part of an HD research team, she emphasizes that she and her coworkers face numerous obstacles. The intense pressure and time constraints that accompany HD research force the researchers to live demanding and hectic lifestyles. Because the lab is part of an HD coalition that meets every six months, as well as other organizations that meet regularly, the Cattaneo lab is expected to maintain constant progress. The labs in the coalition are the best in the world and Dr. Cattaneo said she feels pressure to live up to high expectations. She confesses that she is usually unable to sleep the night before coalition meetings because the excitement and pressure are great.
In order to be able to report new results every six months, members of the lab cannot afford to waste any time. They generally arrive at the lab at 8:30 am and leave between 9:00 pm and 11:00 pm. If they leave at 6 pm, they consider it a half-day. Furthermore, they do not take weekends off. As researcher Chiara Zuccato put it, “There are twenty-four hours in a day – I need to sleep about seven hours and it takes over an hour to get to and from work. For pretty much the rest of the day, I´m in the lab.” Dr. Cattaneo has two children and it is very hard for her to spend so much time away from them. Luckily, it is an Italian way of life to have support from the family and her mother-in-law helps take care of the children.
Many people are not able to handle the pace of this lifestyle, and it is not uncommon for researchers to leave the lab after a year or two. Ironically, Dr. Cattaneo enjoys her work so much that it is hard for her to be away from the lab. The last time she took a vacation she got up very early every morning before her family woke up to check her e-mail and maintain correspondence with the lab.
A big obstacle in Italian academia is the lack of available jobs. Currently, the government is attempting to abolish assistant professorships. If this happens, even individuals with many qualifications will not be able to find positions within academic settings. An individual could be highly prolific for fifteen years and make significant research discoveries and still not be able to advance in the field. This situation in the US would be equivalent to post doctoral students never being able to become professors. Dr. Cattaneo sees this lack of job positions as an obstacle because it will be even harder to recruit new people into her lab.
Because the lab uses stem cells and there are numerous ethical issues surrounding stem cells, it has encountered a few obstacles in the form of religious and political interference. (For a concise discussion on stem cell ethics, click here.) However, members of the lab believe that this problem is a result of the public being misinformed. They believe that once the public better understands stem cell research this issue will no longer be an obstacle.
Another obstacle arises when the researchers must decide whether they should devote to making an old experiment more complete or whether they should devote time to starting a new experiment. On one hand, they want the experiment to be completely thorough. On the other hand, their minds are racing with new ideas and they want to start new experiments. This situation is worsened by the fact that it is difficult for the researchers to satisfy both their curiosities and their own expectations. In one instance, Zuccato had already written and submitted a paper about an experiment when she realized that there was something missing. Despite having a ton of other things to do, she immediately stopped submission of the paper and decided to redo the entire experiment with added conditions to make it more thorough. Thus, for researchers, it is a constant struggle to balance thoroughness with productivity.
Surprisingly, Dr. Cattaneo explains that money is not normally a major obstacle for her lab, because it is supported by many sources, including the University of Milan, the European community, and many American foundations. Happily, for at least the next four years, there is adequate funding. Dr. Cattaneo is very thankful for this: she points out that “research always begets new research,” and funding is crucial for exploring new possibilities.
Conclusion
Between their work on normal huntingtin function, mutant huntingtin function, and the development of stem cells into useful nerve cell models, the members of the Cattaneo lab will have their hands full for many years to come. Dr. Cattaneo knows that she and her fellow lab members will continue working their twelve hour days in hopes of finding new treatments for HD. As Cattaneo puts it, in Italy, the researcher´s reward “is not monetary; it is a dot on a gel” – that is, a single dot showing the successful results of an experiment. For Cattaneo, helping young enthusiastic researchers get started in the field is almost as rewarding. Dr. Cattaneo reminds us that “science has no guarantees but one: that researchers will keep working.” She encourages people affected by HD to learn about the research efforts going on around the world, and she hopes that the knowledge that “someone is thinking about this [disease] every minute of every day” can be comforting and reassuring.
For further reading
- To find out more about the Cattaneo lab, please visit its website at http://users.unimi.it/DPS/struttura.php?id=10.
- For more information about the Huntington´s Disease Society of America and their Coalition for the Cure, visit the HDSA website at www.hdsa.org.
- Cattaneo E., Rigamonti D. Goffredo D. and Zuccato C. (2001) Loss of normal huntingtin function: new developments in Huntington´s disease research. Trends in Neurosciences, 24:3, 182-187.
This article is fairly easy to read. It succinctly summarizes the background of HD as well as the various hypotheses about its molecular pathology.
- Rossi F. and Cattaneo E. (2002) Neural stem cell therapy for neurological diseases: dreams and reality. Nature Reviews Neuroscience, 3, 401-409.
This is a very in-depth article about the therapeutic potential of neural stem cell therapy. It also explains all of the obstacles that must be overcome before neural stem cell therapy can become a viable treatment.
- Zuccato C. et al. (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington´s disease. Science, 293, 493-496.
This is a technical article that describes how the beneficial activity of huntingtin is lost in people with HD and how this leads to decreased production of BDNF.
- Zuccato C., Tartari T., Crotti C., Goffredo D., Valenza M., Conti L., Cataudella T., Leavitt B. R., Hayden M. R.,Timmusk T., Rigamonti D. and Cattaneo E. (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nature Genetics, 35: 76-83.
This article is very technical. It describes in detail how normal huntingtin increases transcription of BDNF by silencing NRSE.
– A. Milczarek and D. McGee, 04/29/05
More

Dr. Steinman’s research has focused on the use of cystamine, a transglutaminase (TGase) inhibitor, as a potential treatment for Huntington’s Disease. Previous studies have shown that HD is associated with the formation of protein clumps, or aggregates, in the brain. Protein aggregation has been linked with some of the neurological symptoms of HD. (To learn more about protein aggregation, click here.) Earlier research has also shown that cystamine inactivates transglutaminase (TGase), an enzyme that helps produce these clumps of huntingtin protein. Therefore, Steinman and his former graduate student, Marcela Karpuj, PhD, reasoned that cystamine might control the disease by preventing the formation of huntingtin protein clumps.
Cystamine has been shown to inhibit the activity of TGase in two ways – one that flips the “off switch” and one that blocks huntingtin from binding. First, cystamine inactivates TGase through a disulfide-exchange reaction, a process by which proteins are activated or inactivated by the folding and unfolding of the amino acid chains that make the proteins. Second, cystamine competitively inhibits TGase. Since cystamine and huntingtin are both capable of binding to the same active site on the TGase enzyme, cystamine will block huntingtin from binding to TGase and thus limit TGase activity.
Steinman and Karpuj tested the effects of cystamine on mice that had a portion of the human HD allele of the Huntington gene (exon 1) inserted into their DNA. They knew that mice with exon 1 exhibited clinical pathologies very similar to the symptoms of human juvenile HD (including the presence of huntingtin protein aggregates and increased TGase activity in the brain), making them good test subjects for the study. (To learn more about juvenile HD, click here.)When cystamine was injected into the mice, TGase activity in the brain was reduced by up to 40% after only ten minutes. To test the effects of cystamine on the clinical symptoms of the HD mice, the drug was administered to mice that clearly demonstrated tremors and abnormal movement. Mice treated with cystamine generally exhibited fewer tremors, decreased abnormal movement, and a lower incidence and delayed onset of HD symptoms. They also lived 20% longer on average.
To the researchers’ surprise, however, cystamine did not reduce the number of huntingtin clumps in the brain. So although their hypothesized clinical outcomes were realized, their hypothetical mechanism was not. This unexpected finding led the Steinman lab to search for an explanation of the alleviated tremors and prolonged life. They started by looking for differences in gene expression between the mice that were treated with cystamine and the mice that were not. The researchers found two genes that were expressed much more in the cystamine-treated mice. A third, related gene showed increased activity in human HD patients, although not in the mice. All three genes code for proteins that are known to protect brain cells from damage. Therefore, cystamine seems to boost the levels of the brain’s natural protective proteins.
Although these findings suggest that cystamine could be a new treatment for HD, research for even more effective and specific drugs continues. Steinman suggests that a combination of cystamine and other drugs may have even greater benefits.
For more information on the research done by Lawrence Steinman, visit the following website: http://steinmanlab.stanford.edu/
For Further Reading:
[toc]
-J. Czaja 8-28-02
More
HOPES interview notes
Dr. Ron Kopito and Dr. Brigit Riley
April 5, 2006
In April 2006, Christina Chen and Justine Seidenfeld visited the Kopito Laboratory in the Department of Biological Sciences at Stanford University. HOPES would like to thank Dr. Ron Kopito and the members of his lab, particularly Dr. Brigit Riley, for taking time out of their busy schedules to meet with us, and share their perspectives on Huntington’s Disease research.
Introduction
In a cell, proteins are constantly being produced or destroyed in order to ensure that a healthy balance is sustained. Many neurodegenerative diseases are associated with an imbalance of proteins. They often involve mutated or misfolded proteins that are toxic to the nerve cell. The Kopito lab looks at one of the most well-known systems for clearing out misfolded proteins, the proteosome. The proteosome is a protein complex that digests unwanted proteins into short chains of amino acids. The proteosome has many roles in the cell and plays a part in many processes, such as the cell cycle, communication between proteins, and the production of proteins. Research about the proteosome is an increasingly important part of our understanding of disorders involving misfolded proteins like HD.
Becoming involved in HD research
When asked how he became interested in HD research, Dr. Kopito acknowledges that he has a different background than most scientists currently studying the disorder. He was originally interested in researching Cystic Fibrosis (CF)- a genetic disorder that, like HD, is associated with misfolded proteins. The most common symptom of CF is difficult breathing, which is caused by lung infections that can be treated (but not cured) by antibiotics. There are other symptoms, including sinus infections, poor growth, diarrhea, and infertility.
A study using a model of CF demonstrated that, by impairing the proteosome, mutant CFTR proteins were not broken down and recycled. Instead, they tended to form protein aggregates. This finding indicates that the proteosome plays a key role in delaying or preventing diseases involving misfolded proteins, like CF and HD. If something goes wrong with degradation, misfolded proteins accumulate in the cell and cause problems.
If the proteosome becomes impaired, misfolded proteins tend to form concentrated clusters of proteins aggregates known as inclusion bodies (IBs). IBs were initially discovered in experiments in the Kopito Lab. At first researchers did not know if IBs occurred naturally in a diseased cell. It was possible that they only appeared because of laboratory conditions used for the experiment. To make sure that IBs occurred in the normal course of a disease involving misfolded proteins, Dr. Kopito and his coworkers looked at other diseases that were similar to CF. He wanted to see if these disease also involve the formation of IBs from mutated, misfolded proteins. Kopito’s group discovered that in addition to CF, most neurodegenerative diseases involve proteins that misfold and form IBs. So, the Kopito lab concluded that IBs were a real part of diseases involving protein misfolding.
Next, they looked closer at the IBs to see what other kinds of proteins were found in addition to the misfolded disease-related protein (i.e. huntingtin or CFTR). They found that another protein called ubiquitin (Ub) was always present. Ub is one of the proteins in the proteosome protein complex. If Ub is found trapped in IBs, it means that Ub cannot work with the proteosome to degrade proteins. Therefore, the proteosome cannot function properly. In this case, proteins in the cell that need to be degraded and cleared (like altered huntingtin) will not be. These proteins can then interfere with other parts of the cell that would otherwise be functioning properly.
Getting involved in the HD community
For some time, the Kopito lab was very interested in studying IBs in general. They were not interested in how IBs were related to specific diseases (like HD), since they were found in so many different genetic disorders. Rather, the lab was using lots of different disease models to study IBs- from Cystic Fibrosis, to HD, and more. But in 1998, the Huntington’s Disease Society of America (HDSA) asked Dr. Kopito to give a talk at a conference on his work with IBs. He went to several of their meetings, met many researchers and patients who were part of the HD community, and found the HDSA to be a “cohesive, vibrant organization”. Dr. Kopito says that the HD community and the HDSA are remarkably well-organized and mobilized, much more so than many organizations involved in advocacy for other disorders.
At these HDSA meetings he saw that many people, scientists and non-scientists alike, attended and participated. The HDSA gives grants for research, and encourages scientists to become involved in the advocacy and politics of HD. When he saw how patients, their families, and advocates supported scientists involved with HD research, Dr. Kopito became very motivated; a few years later, he decided to concentrate on HD more seriously. He has been focusing his research on the role of IBs and the proteosome in HD ever since.
The “top-down” approach
On a basic level, it is known that HD is caused by a mutation in huntingtin that causes it to gain a new toxic function and harm the cell. But, as Dr. Kopito explains, there is not a single, direct route from the genetic mutation to the disease. Rather, the mutated huntingtin protein has different effects all over the cell. Dr. Kopito feels that when studying HD, it is hard to start by isolating each of the possible small problems that may be caused by the mutated huntingtin protein. Starting from the endpoint of the disease cascade will not provide much insight into understanding the disease. Rather, you have to study the disease as a whole to gain insight into each of the small steps, and how they interact with one another. This approach would be considered a top-down approach. So, Dr. Kopito likes to use animal models like mice to study the disease, rather than looking at HD only in tissue culture or in vitro. That way, he can look at the whole disease in the context of an animal, rather than just a fraction of it in the context of a test tube or Petri dish.
Current research projects
Dr. Kopito mentions two current projects in his lab that he finds particularly exciting at the moment. The first project involves looking at the timing and appearance of inclusion bodies in HD. Dr. Kopito says he believes that to understand how IBs play a role in HD, he first has to know at what time point they appear over the course of the disease. When Ub accumulates and is incorporated into IBs, it indicates that something is wrong with the proteosome. Dr. Kopito thinks that this happens because at some point over the course of the disease, the proteosome just stops working. All of the extra Ub that should have been degraded needs somewhere to go, and it gets incorporated into an IB. But for Dr. Kopito, the question is when does that occur? When does the proteosome stop working? For more on the proteosome, UB, and HD click here and here.
To answer this question, the Kopito lab is using a model system with mice that have HD. They take samples from the brain tissues of these mice over time and test how well the proteosome is functioning at each time point in order to find the time when the proteosome stops working. Dr. Kopito says that the data from this experiment is forthcoming, and so they should have results to further investigate. He is pleased that he will be able to tell if his pet hypothesis (that the proteosome shuts down early and is important in the onset of HD) is right or wrong. Either way, it will give him a great direction in which to move his research.
Dr. Kopito’s second project is in partnership with a local biotechnology company. Together, they are trying to find biomarkers for HD. A biomarker for HD would be a type of molecule that indicates when neurodegeneration has started. A good marker could be a molecule that has a recognizable change in concentration or structure when neurodegeneration begins. It would be nice to have a biomarker for HD so that you do not have to wait for the motor, behavioral, or cognitive symptoms to appear to know when neurodegeneration has begun. (For more on the symptoms of HD, click here). Biomarkers would allow doctors and scientists to intervene in the course of the disease early on, before it is too late. Ideally, if you could test a blood sample and find a molecule that indicated that neurodegeneration had begun, it would be an important step in being able to test drugs and therapies.
This second project is actually closely related to the first project. Dr. Kopito hopes that the proteosome, or a piece of the proteosome, might be a good biomarker molecule for HD. If the experiments described for the first project prove that the proteosome stops functioning early in course of the disease, it could serve to indicate when it is time to intervene before the motor, behavioral, or cognitive symptoms set in. However, right now, the only ways to test how well the proteosome functions is by doing a biopsy, which is a complicated and very invasive procedure. Dr. Kopito proposes that a cerebral spinal tap might be an alternative, simpler option to test how well the proteosome is functioning.
Autophagy, or “self-eating”
While Dr. Kopito’s lab focuses mainly on the role of the proteosome in HD, some members of his lab look at other mechanisms that the cell uses to break down and clear out unwanted proteins. Christina and Justine met with Dr. Brigit Riley, a post-doctoral student in the Kopito lab who studies a process known as autophagy, and how it is involved in HD. Autophagy literally means “self-eating”. In autophagy, the cell’s membrane encircles organelles, proteins, and parts of the cytoplasm into spheres called vesicles. These vesicles are sent to a part of the cell called the lysosome to release their contents. Then, proteins in the lysosome degrade the contents of the vesicles . See Figure 1 below). Both the proteosome and autophagy are methods the cell has to recycle proteins. But, there are some important differences between the two processes. While the proteosome can selectively target certain proteins that are supposed to live for a short period of time, autophagy is used to digest large organelles and long-lived proteins, and it cannot select for certain proteins.

Figure 1
Autophagy has many purposes in the cell. During development, it helps the cell take up amino acids. If the cell is starving and does not have enough nutrients or energy, autophagy is used to recycle unnecessary proteins to make more important proteins. Autophagy can also be used to defend the cell against invading bacteria or viruses. It is also thought to play a protective role against the progression of human diseases like cancer, Alzheimer’s disease, Parkinson’s disease, and HD. (For more on other neurodegenerative and related diseases, click here.
Autophagy and HD
There have been many scientific models recently developed to look at the role of autophagy in HD. Scientists think that autophagy is triggered to help protect the cell when the proteosome system is overwhelmed by too much aggregated protein. That is, the proteosome is the first line of defense, whereas autophagy is the second line of defense against altered huntingtin protein.
In this case, the proteosome can deal with small huntingtin aggregates by selectively targeting them for degradation. However, when there are too many of these misfolded proteins, the system gets overwhelmed. Then, the excess of misfolded proteins are shuttled along the microtubules (which are the tracks that allow protein to move across the cell) to form clumps of misfolded proteins called aggresomes (the name for IBs found in the cytoplasm as opposed to in the nucleus of the cell). Logically, the cell would form aggresomes as protection because it is much easier to autophage large clusters of altered huntingtin proteins than small proteins floating all over the cell.
However, there are still a lot of questions surrounding this process. We do not know if only the aggresomes are degraded by autophagy. It could be that any altered huntingtin proteins that are not incorporated into aggresomes are also autophaged. We also don’t know if the molecular signals that activate autophagy to degrade aggresomes also trigger autophagy for other purposes, like cell development or cell starvation.
Autophagy research in the Kopito Lab
Dr. Riley looks specifically at the role of the microtubules in triggering autophagy. Microtubules may assist the fusion of the vesicles and the lysosome to form the active autolysosome that carries out autophagy. Dr. Riley believes that this is the link between microtubules and autophagy. In fact, experiments have shown that mutating the microtubules to disrupt their structure interferes with autophagy.
Researchers do not know for sure that autophagy is completely protective for the cell. It is possible that aggresomes activate autophagy, but instead of only targeting the aggresomes, autophagy targets everything in the cell. In this case, autophagy would be blindly degrading essential proteins and organelles along with the aggresomes, thus harming the cell. Autophagy would be a good temporary solution, but bad for the cell in the long run.
Dr. Riley also suggests that when aggresomes get degraded during autophagy, the resulting fragments of altered huntingtin protein act as the toxic molecule that harms the cell. Another possibility is that the aggresomes clog the lysosome, preventing it from acting normally in other parts of the cell. As she says, there are a lot of options to explain how autophagy functions in HD. However, autophagy research is relatively new, so a lot remains to be done.
There are a lot of experimental models used in the lab, and Dr. Riley mentions a specific tissue culture cell line that comes from mouse nerve cells. These cells have the altered huntingtin protein, and it is under the control of a “conditional promoter”. By adding or taking away a certain chemical called tetracycline, researchers can turn the production of the altered huntingtin protein either on or off at will. This is a very powerful model system because it allows you to produce enough altered huntingtin protein for HD to begin. Then, you can turn off production of the protein, and look at how autophagy is clearing it out of the cell. Dr. Riley particularly likes working with cell lines like these. She thinks it is a good model system for preliminary studies before taking her findings and applying them to more complex models, such as mice. But she has a few concerns. She recognizes that mice nerve cells do not behave the same way as human nerve cells, so she wonders if findings from model systems like cell lines and mice will apply to humans well.
More on Dr. Riley
So how did Dr. Riley become interested in studying HD? As an undergraduate, she started as an organic chemist, working in a lab that studies nitric oxide synthase in Parkinson’s and Alzheimer’s disease. Both of these are neurodegenerative diseases that have many similarities to HD. At the time, she was also volunteering in a local nursing home, and began to interact with patients with the very same neurodegenerative disorders she was studying in the lab. As a graduate student, she decided to study biochemistry. She began looking at the role of the proteosome in spinocerebellar ataxia, which is a neurodegenerative disorder associated with expanded polyglutamine chains, like HD. Specifically, she was looking at a set of proteins that acted as an alternative to ubiquitin. This line of inquiry led to her interest in autophagy as an alternative method of recycling proteins compared to the well-studied proteosome system. Since then, Dr. Riley has switched model systems and is now studying HD, but she plans to always stay in the area of science devoted to studying neurodegenerative disorders.
Who does Dr. Riley find to be an inspirational scientist? She first identifies Dr. Yee Yamamoto, who created the first tetracycline-regulatable model system for an HD mouse. This cell line allows scientists to control when the altered huntingtin protein is produced in the mice, and when to turn off its production. Many other scientists applied this idea to other model systems, including the tissue culture cell line that Dr. Riley mentioned using earlier.
Life as a post-doctoral student
Dr. Riley came to the Kopito lab as a post-doctoral fellow. She entered the lab with a few other scientists who were interested in working on autophagy, so she had a new group of people to collaborate with on projects. She found that the Kopito lab is unique because it has a good environment and good energy. Everyone works together, especially within the group of graduate students and post-doctoral fellows working on autophagy. She also notes they are unusually dynamic, and they like to act on an idea quickly rather than wait around. She also comments that Dr. Kopito is great about helping his graduate students and post-docs to act quickly on a project idea by supplying them with necessary equipment, materials, and advice.
As a post-doctoral student, Dr. Riley finds that it is a challenge to balance bench work and experiments with meetings, writing papers, meeting people, and making contacts, all of which are important parts of a post-doctoral student’s work. She found that it was not as hard to balance responsibilities as a graduate student, but there are more tasks and more pressure now. She also wants to stay on top of current research about HD and autophagy, but finds it more difficult than she expected. Because there are so many different approaches to the subject, it is hard to know what is going on in the field. Furthermore, the lack of communication between other scientists in the field makes it hard to design experiments.
When asked if her fellow researchers at the Kopito lab have focused their goals on contributing to a cure for HD, or rather on the joy of scientific discovery, she says the Kopito lab is a combination of attitudes. Her old lab (where she did her graduate studies) focused on either curing or understanding the initial steps of HD. At the Kopito lab, there is more of a balance between both attitudes. Personally, when she thinks only about the joy of discovery or the intellectual pursuit of understanding HD, she becomes concerned that it isn’t really relevant to the people who are affected by HD. She thinks that scientists are really far removed from the fact that this is a fascinating scientific question, but also involves patients and their families.
For Dr. Riley, the excitement in studying autophagy is in the hopes of “find[ing] a drug that would promote it”. But it also prompts questions about the nature of HD treatment- do people want to take a drug treatment for their whole lives? Is it too inconvenient or difficult of a method? Optimistically, she says, maybe this drug approach will be used to treat symptoms starting in 5 to 10 years, but it would be nice if gene therapy was possible. If they were developed around the same time, it would give people options in how they want to be treated, which she believes is an important aspect of scientific research and medicine.
Dr. Kopito, HD research, and the public
When asked what the most rewarding parts of research are to him, Dr. Kopito stressed that he most enjoys the satisfaction of discovering and understanding something new. In his role as a professor and researcher, he is also a mentor to the graduate students and post-doctoral students in his lab. He emphasizes how much he enjoys teaching and nurturing other people’s sense of discovery. But Dr. Kopito further elaborates by saying that his research program is not intended to directly cure HD, but rather to further understand the background and the cause of the disease. As he explains, he finds HD to be a “compelling medical problem, an interesting scientific problem, and really, a human problem”. But when asked if he hopes he is making an impact on lives, he recognizes that his role is indirect and not concrete. While he feels he is not directly accountable to HD patients, he also recognizes that scientists have to be responsible to the HD community. Scientists have to spend any grant money on research that will get scientists or doctors closer to a place where they can find a treatment.
This line of questioning raises many ethical questions for Dr. Kopito. He wonders- what if it turns out that inclusion bodies are not the problem to target in HD? Can he still work on studying IBs in other diseases, because he finds it to be interesting and compelling research? Or would it be more ethical to switch the focus of his research, and look at other mechanisms that may be involved in causing HD? He thinks about these questions a lot, meeting with other people to talk about inspiring new ideas within the group of researchers working on HD.
Finally, Dr. Kopito emphasizes that the biggest myth about research is that it proceeds in a very dry, set manner from hypothesis to experiment to truth. The public does not often get to see how qualities like intuition, passion, and creativity are involved in conducting research. If scientists were to fit the cold, objective stereotype, there would be no important discoveries. Rather, when scientists get ideas, some people see it as crazy risks, but they have to have enough faith in their own ideas to work on it, and they have to convince other fellow scientists and students to take risks along with them. Scientists have to design an experiment with objectivity and creativity. An experiment must be designed to test, not prove, a hypothesis- scientists must be willing to acknowledge when they are wrong or their hypothesis is incorrect. But, that can be hard because the ego gets wrapped up in it. Throughout their careers, scientists have to utilize ego, passion, commitment, and risk to conduct experiments. For Dr. Kopito, research is a vital, dynamic profession.
For further reading
- Find the Kopito Lab at: http://www.stanford.edu/group/kopito/
- Debnath, J, et al. “Does Autophagy Contribute to Cell Death?” Autophagy 2005. 1 (2): 66-74.
A good review of autophagy, not too technical language. Discusses roles of autophagy in both cell death and cell survival.
- Ross, CA, et al. “What is the role of protein aggregation in neurodegenation?” Nature Reviews Molecular Cell Biology 2005. (11):891-8.
A more complex review of disease involving protein aggregation, inclusion bodies, and the roles of both in causing diseases or acting as a protective response.
- Yorimitsu T, et al. “Autophagy: molecular machinery for self-eating.” Cell Death Differ.2005. 12 (2):1542-52.
A technical paper on autophagy, how it works, and the various roles it plays in the cell.
- Iwata, A, et al. “HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin.” J Biol Chem. 2005. 280(48):40282-92.
A technical research paper on the role of microtubules on forming aggregates and how they participate in forming vesicles and activating the lysosome.
- Levine, B, et al. “Development by self-digestion: molecular mechanisms and biological functions of autophagy.” Dev Cell. 2004. 6(4):463-77.
A good review of autophagy that goes into a bit more detail about the sets of genes involved in autophagy and different analogous genes in model organisms.
- Kopito RR. “Aggresomes, inclusion bodies and protein aggregation.” Trends Cell Biol. 2000. 10 (12):524-30.
A review that clarifies the similarities, differences, and relationship between aggregates, IBs, and aggresomes.
More
In December of 2004, HOPES team members Kim Taub and Jia Hou visited Washington University in St. Louis, Missouri to get an inside view of an HDSA Center of Excellence. They met with Melinda Kavanaugh, a social worker in the Department of Neurology Movement Disorders Section, and Dr. Samer Tabbal, a neurologist.
Washington University Medical Center has been named a Center of Excellence by the Huntington’s Disease Society of America (HDSA). The HDSA recognizes those centers that provide excellent care for people with HD, participates in community outreach, and conduct research and clinical trials. Washington University serves more than the people of St. Louis; it reaches out to much of the Midwest.
Caring for HD
The Center consists of a team of specialists, including several neurologists, a neuropsychiatrist, a social worker, a genetic counselor, and an occupational therapist. It is important that they approach their patients as a team because of the complicated nature of HD; people with HD often have several distinct medical complications that require specialized attention. For example, a neurologist may be able to help with choreonic (jerky, uncontrollable) movements, while a psychiatrist or licensed clinical social worker can assist in coping with potential psychological issues. A patient may also want to talk to a genetic counselor if he or she is considering having children. No single person is qualified to deal with all the facets of this disease. Consequently, it is best to approach and treat people with HD using a team of specialists who work together.
Melinda Kavanaugh, the Center’s social worker, met with Taub and Hou to explain how things work at Washington University. The team sees patients every Wednesday. Since the Center is located in the Movement Disorders section of the Department of Neurology, the team sees not only people with HD, but also people who have other diseases resulting in motor problems. Overall, the team sees about 2 to 8 people with HD each week. When patients come in, they first see Melinda to undergo cognitive testing using the Unified Huntington’s Disease Rating Scale (UHDRS).
Melinda made an important observation that is perhaps overlooked by the general population: “Primarily, I think this is a psychiatric disorder.” She explained that, most of the time, people with HD are not even aware of the erratic movements that they have. Instead, the things about the disease that bother them most include changes in mood, depression, the inability to control anger, and the frustrations arising from not being able to work. Because patients seem to be bothered most by mental as opposed to physical problems, the Center team focuses primarily on psychiatric and psychosocial aspects of HD. (Psychosocial is just what it sounds like; it involves how psychological aspects can affect social aspects of one’s life and vice-versa.) Rather than treating the movements right away, which they feel has historically been a huge mistake, the team instead focuses on family involvement, communication, and starting a chain of advocacy. They emphasize the importance of establishing good communication to prevent divorce and abandonment, which occur when family members do not understand their loved ones’ condition.
After seeing Melinda, the patients see Dr. Tabbal, a neurologist, for evaluations and referrals to occupational, physical, and speech therapists. Because HD can have a profound effect on a person, team members always recommend that their patients see a general therapist as well.
Community Outreach
Besides seeing patients, the Center is especially active in reaching out to the greater HD community. Since the Center does not have its own HD facility, the team must attempt to reach “outside the clinic.” It is very difficult to find the type of specialized care offered at Washington University in the surrounding rural areas. Melinda and her coworkers therefore focus on extending care and education to these underserved areas. They travel to nursing homes to teach caregivers about HD and how to more effectively care for their patients. They also provide family and community outreach by placing ads in local papers and on local radio stations in rural areas, as well as by creating support groups to target these otherwise isolated populations.
Melinda explained that Dr. Joel Perlmutter, the Center Director, recognizes that the main impact of HD is psychosocial, that it can impede the socioeconomic development of families with this disease. This problem is especially devastating in farming communities with a tradition of large families. Large family sizes may result in concentrated clusters of HD, such as those found in southern Illinois.
Research at Washington University
An important part of what makes the Washington University Medical Center a Center of Excellence is its participation in research on HD and clinical trials. Center Director Dr. Perlmutter has conducted much research on movement disorders. Dr. Perlmutter has published at least 100 articles in the past 20 years, the majority of which relate to Parkinson’s disease. (For more information on how Parkinson’s and HD are related, click here.)
Patients who come to the Center also have the opportunity to participate in national clinical trials. Two recent clinical trials are PREDICT-HD and the OX-Phos Study. PREDICT-HD is an observational study that tracks the progression of people with HD over the course of four years. When the participants come in for their visits, they undergo cognitive testing, MRI (Magnetic Resonance Imaging) scans of the brain, and a motor evaluation. The goal of this study is to find neurobiological predictors of HD; they are looking for some readily identifiable sign in the brain that will indicate the progression of HD.
The OX-Phos Study is named for oxidation-phosphorylation, a very important step in cellular energy production. This study examines people who have tested positive and negative for HD, but have not started to have symptoms or only have very mild symptoms. This study involves one or two patient visits to compare how people with HD use sugar and oxygen in the brain differently from people who don’t have HD. This study is important because it is known that the altered huntingtin protein causes problems with energy metabolism. By studying how oxygen and sugar (important ingredients in creating usable energy in the body) are used in the brains of people with HD, researchers can better identify metabolic problems and develop treatments to address them. (For more information of energy metabolism and HD, click here.)
Neurology at Washington University
There are 400 people who work in the neurology department at Washington University. Dr. Tabbal, one of 40 clinicians at the Center, is also founder of the first HD clinic at the University of Arkansas. He is proud of the university’s prestige in the field: Washington University receives between 6 and 7 percent of the annual funding given by the National Institutes of Health (NIH), or roughly 22-23 million dollars.
Remarkably, the Positron Emission Tomography (PET) scanner was invented at Washington University. Although the original scanners were enormous, the ones used today are only slightly larger than the subjects that go in them, usually people and larger animals. PET scanners are a major research tool at the Center, something that distinguishes Washington University from other research labs.
Before PET was invented, researchers could only infer information about the brain through dissections after death and animal studies. PET allows researchers to track the function of the brain of a living, alert subject. To do this, a very small amount of a radioactive substance that is similar to a substance already found in the body is injected. As any radioactive substance decays, it emits particles called positrons. The scanner then picks up these positrons as the radioactive marker in the subject’s brain decays.
The specific kind of marker used depends on the area or function of the brain being studied. For example, a researcher studying glucose metabolism in the brain will inject a substance similar to a normal glucose molecule, but with an added radioactive fluorine atom, called TRIUMF. To study blood flow to various areas of the brain, radioactive water or 6-F-dopa is injected. The amount of radiation a subject is exposed to is typically less than he or she would be exposed to during an X-ray. Unlike X-rays, however, which produce only a shadow image, PET scans allow doctors and researchers to track changes in the subject’s brain as various cognitive tasks are performed.
Information gained from PET scans and genetic testing have contributed immensely to the growing body of knowledge on HD. Since 1992, when genetic testing became available for HD, scientists have been “learning exponentially” about the condition. Dr. Tabbal cautions that this knowledge must be used wisely.
“[Pharmaceutical companies] have a library of compounds,” says Dr. Tabbal, “Many are not FDA regulated. They are also expensive and ineffective, and we have little knowledge of their long-term effects. The difference between Western medicine and sorcery is that with Western medicine, we do research and experimentation to find out if there is validity to a drug’s proclaimed effect. Unfortunately, most of these drugs are more witchcraft than medicine.”
A drug that has recently gained publicity is Coenzyme Q-10. Q-10 supposedly prevents changes in nerve cells’ oxidation-phosphorylation, a normal metabolic process. These changes are believed to contribute to apoptosis of nerve cells, or nerve cell death. In clinical trials, only doses of 1200 mg have been shown to have any marked effect on persons with HD. Even then, the benefits are few. Dr. Tabbal points out that many people still pay a lot of money for doses of 50-100 mg.
According to Dr. Tabbal, the research conducted at the Washington University neurology department focuses on phenomenology, which means putting a great emphasis on understanding the process of disease rather than merely recording results. Otherwise, treatment becomes a shot in the dark.
Dr. Tabbal jokes, “There are more supporting staff [like Melinda] than doctors in the Movement Disorders department.” This high ratio of supporting staff to doctors is for a good reason. Like Melinda, Dr. Tabbal believes that HD is mainly a psychiatric disease which requires counseling as much as, if not more than, medical intervention to treat.
Dr. Tabbal is a friendly, upbeat doctor who has come close to experiencing the effects of HD firsthand. Every summer, he participates in a five-day camp for people with HD at Columbia University. Everything at the camp caters to people living with HD: the cabins are ergonomically designed and the daily schedule is highly structured and detailed.
“When you are surrounded by people with Huntington’s, you become the odd one,” Dr. Tabbal says. “It is the only way you can really understand what’s it’s like. You understand why it takes so long to dress, how they fall.”
Dr. Tabbal believes that a good support system, physical therapy, relevant medicines, and the right attitude should be the foundation of treatment. He believes that there is hope after a diagnosis of HD. People at risk for HD should not be afraid of what the doctor might say because, after all, the diagnosis is the first step in treatment. Dr. Tabbal adds his final piece of advice with a smile, “Make sure you see a good doctor.”
For further reading
- To find out more about HDSA Centers of Excellence, visit http://hdsa.org/about-hdsa/centers-of-excellence/.
- To find out more about the Washington University Center of Excellence, visit the website at https://neuro.wustl.edu/patient-care/movement-disorders-2/huntingtons-disease/.
- To find out more about PREDICT-HD, visit http://clinicaltrials.gov/ct/show/NCT00051324?order=1.
More
Gene therapy may switch off Huntington's
By Bob Holmes, Banff
Using gene therapy to switch off genes instead of adding new ones could slow down or prevent the fatal brain disorder Huntington's disease. The method, which exploits a mechanism called RNA interference, might also help treat a wide range of other inherited diseases.

"When I first heard of this work, it just took my breath away," says Nancy Wexler of Columbia University Medical School, who is president of the Hereditary Disease Foundation in New York. Though the gene-silencing technique has yet to be tried in people, she says it is the most promising potential treatment so far for Huntington's.
It involves a natural defence mechanism against viruses, in which short pieces of double-stranded RNA (short interfering RNAs, or siRNAs) trigger the degradation of any other RNA in the cell with a matching sequence. If an siRNA is chosen to match the RNA copied from a particular gene, it will stop production of the protein the gene codes for (see graphic).
Huntington's is caused by mutations in the huntingtin gene. The resulting defective protein forms large clumps that gradually kill off part of the brain. Studies in mice have shown that reducing production of the defective protein can slow down the disease, and Beverly Davidson at the University of Iowa thinks the same could be true in people.
"If you reduce levels of the toxic protein even modestly, we believe you'll have a significant impact," she says. Late in 2002, her team showed that it is possible to reduce the amount of a similar protein by up to 90 per cent, by adding DNA that codes for an siRNA to rodent cells engineered to produce the protein.
Disease-causing genes
The team was the first to use gene therapy to deliver such a payload, and they have now done the same with the huntingtin protein itself. Completely silencing the gene in people with the disease is not an option because brain cells may not survive without the protein. But we have two copies of most genes, and usually only one is defective in people with Huntington's.
Working on a similar disease using human cells, Davidson and her colleague Henry Paulson have now shown you can make an siRNA that recognises and silences only the mutant gene.
They could not target the disease-causing mutation itself because, as in Huntington's, the mutation merely makes a long stretch of repeats even longer, without actually altering any particular short sequence. But they did find another difference, a change in a single DNA letter that appears in 70 per cent of defective genes.
Adding an siRNA that matches this telltale sequence reduced expression of the defective protein by over 80 per cent, while production of the normal protein was hardly affected, Davidson told a gene therapy conference in Banff, Canada, last week. The hunt is now on for similar mutations in the huntingtin gene itself. One promising candidate has been discovered in about 40 per cent of disease-causing genes.
The same approach could probably be used for many other genetic disorders. Even if both copies of a gene are faulty, a healthy copy of the gene could be added alongside an siRNA that turns off both defective copies.
Related articles
RNAi protects living animals against disease
Interference technique is clean cancer killer
Clue to treatments for fatal Huntington's
Source:
[toc]
More
In April 2007, HOPES researcher Justine Seidenfeld visited the Baltimore Huntington’s Disease Center (BHDC) in the Department of Psychiatry at the Johns Hopkins University School of Medicine in Baltimore, MD. The BHDC has been designated as a Center of Excellence by the Huntington’s Disease Society of America (HDSA). Centers of Excellence are recognized not only for the quality and scope of care they provide for their patients, but also for their clinical and basic research efforts, and the outreach services they provide to the general community. For more on the HDSA’s Center of Excellence program, please click here. The Baltimore Huntington’s Disease Center serves most of the mid-Atlantic region, providing services to Maryland, Pennsylvania, Delaware, New Jersey, Washington DC, and Virginia.
HOPES would like to thank the Baltimore Huntington’s Disease Center, particularly Mr. Abhijit Agarwal, Ms. Kit McFarland, Ms. Debbie Pollard, and the director of the Center, Dr. Chris Ross, for taking time out of their busy schedules to speak with us and share their perspectives on Huntington’s disease research.
Clinical Programs
The Baltimore Huntington’s Disease Center is the only HDSA Center of Excellence based in a psychiatry department (instead of a neurology department) of a medical school or hospital. As such, the Center can pay special and expert attention to treating the psychiatric and psychosocial aspects of the disease, for which we now often have effective symptomatic treatment. However research focuses mostly on brain imaging, neurobiology, genetic mouse models and other basic science, and clinical trials of new therapeutic agents. For more information on the symptoms of HD, click here.
Ms. Debbie Pollard is the clinic coordinator at the BHDC, and she helps patients when they first come to the Center. Ms. Pollard performs full physical exams for each patient to initially determine what symptoms they have, takes a full family and personal health history, and helps to generally guide patients as to how the center works and what services they provide.
The Center runs two separate multidisciplinary clinical programs to provide services to incoming HD patients. Additionally, there are related social services (such as community resource referrals and housing assistance) available in both programs. Confidentiality is strictly upheld in all cases and practices.
One of the clinical programs is an “Evaluation and Research” program, which runs every Tuesday from 1-4pm. The program is over 25 years old, and is supported by the HDSA and the National Institutes of Health (NIH). Thanks to these research grants, all of the services are (presently) provided with no charge. Patients can either make an appointment to come in on their own, or they may be referred to the Center by other physicians or neurologists in the area. At the first meeting, a number of neurological and psychiatric evaluative tests are conducted in addition to the detailed medical and family history that Ms. Pollard takes. Individuals who do not know if they have the altered huntingtin gene, but are presenting some of the symptoms of HD, can take a genetic test for HD (or other potential neurodegenerative disorders) if they so wish. Other staff members of the BHDC may see the patients for follow up services like consultations, case management (including symptomatic treatments for chorea or anti-psychotics if appropriate), counseling, and referrals if it is necessary.
The second clinical program offered at the Baltimore Huntington’s Disease Center is the “Continuing Care Program”. This program offers ongoing medical care for HD patients, as well as counseling and social services for them and their families. The primary purpose of the clinic is to treat and manage HD symptoms through medication, counseling, and to help patients to adjust their environmental surroundings. Patients can be referred to see staff members at the center who serve as neuro-psychiatrist specialists, social workers, or be referred for occupational therapy and speech therapy. The goal of the staff is to work with patients and their families to develop a tailored program for care. As opposed to the “Research and Evaluation” program, services from this program must be paid for, but health insurance is accepted when it applies. However, a generous grant from the HDSA allows the BHDC to charge for services based on the patient’s ability to pay and provide services for free to individuals that cannot otherwise afford it.
The clinic also conducts genetic tests for family members of those with HD who want to know their gene status. Genetic testing is a serious issue, and should only be undertaken after much consideration and counseling- for more information about genetic testing for Huntington’s Disease, please click here. They also offer counseling to many asymptomatic individuals who have the altered HD gene, in order to help them adjust to the kinds of lifestyle changes they can anticipate when symptoms begin to appear. Ms. Pollard emphasizes that as a Center of Excellence, the clinic is open to anyone regardless of the ability to pay, so anyone may come in for these services.
Social Services
Social worker Ms. Kit McFarland explains that the BHDC is mostly a research operation that also provides care to patients. A large part of Ms. McFarland’s job is to provide tailored information about HD to families or individuals who have specific concerns. She also puts together an initial package of basic information for those who are unfamiliar with HD to use before they need more specialized help. Much of the time, the people who come to her for advice are families who have just learned that HD runs in their family (usually after one member was diagnosed with it) and they desire more information.
Ms. McFarland feels her job as a clinical social worker is to generally help people learn how to best support and stay involved with their family members that have been diagnosed with HD. For example, she gives them tips to help them make certain that the member of the family with HD stays well-nourished, such as using Ensure shakes. For family members who act as caregivers, she often finds that it is difficult for them to understand how their loved one will change behaviorally throughout the course of the disease, and so she tries to provide ways for them to cope with this issue. They may commonly encounter violent reactions from the family member with HD, but sometimes it can be hard to tell if it is intended violence or a motion from chorea. For those afflicted with HD, one of the most difficult parts of the disease is the gradual loss of their freedom- particularly when in comes to driving. If it comes time for the HD patient to stop driving but they refuse, Ms. McFarland might tell the family members or caregiver that the patient needs to be taken for a driving test, and tries to get the family involved in this decision. For more on HD and driving, please click here.
Another one of Ms. McFarland’s chief tasks is to provide help to the outside community, particularly those who have reached the stage of HD where they need some kind of assisted living arrangement- either in nursing homes or other facilities. This is a difficult process because many individuals with HD are often not prepared to relinquish their independence, even when it is necessary. She also works directly with employees of nursing homes and assisted living facilities in the area to teach them skills they need to serve residents with HD. She explains that many of the issues that nursing home or assisted living facility employees will encounter with their HD residents are actually very common among individuals of that age- such as problems with getting residents to eat. The major differences in residents with HD, that pose greater problems to the staff, are very rapid weight loss and aggression. For more on the manifestations of aggression in HD, click here. Ms. McFarland is also highly involved in much of the Center’s efforts to provide help, information, and services to those patients who have difficulties with transportation and cannot come to the center themselves.
Clinical Trials
Mr. Abhi Agarwal is the clinical research coordinator for the BHDC, and his job is to organize the enrollment and participation of patients in various clinical trials run through the Center. As Mr. Agarwal explains, the Center offers a wide range of opportunities for individuals and their families to participate in a variety of clinical research studies. Participation in these research trials is free of charge, thanks to a number of research grants from the National Institutes of Health (NIH), the Huntington’s Disease Society of America (HDSA), and the Huntington’s Disease Foundation (HDF).
Mr. Agarwal explains that they take a diverse array of approaches to looking at HD, conducting clinical studies that involve genetics, neuropathy, autopsy-based studies, fMRI studies, and basic research studies. He elaborates further on the clinical research studies currently being conducted at the Center:
1) The Longitudinal Core Study: This project is an observational study, which means that it is not intended to test potential treatments for HD, but rather is a study that will help advance our understanding of the natural onset and progression of HD. To enroll in the trial, the only requirement is to have undergone testing for the altered HD gene- participants don’t need to actually have HD, because control subjects are needed as well. Any patient that participates in the study at the BHDC is seen annually from their enrollment until their death, and at each yearly meeting they receive full neurological and psychological evaluations to assess the how far along motor, cognitive, and behavioral symptoms have progressed. They also receive functional magnetic resonance imaging, or fMRI scans on every other visit to look for structural differences in the brain over time (changes in the shape of the brain, usually related to nerve cell death), and to compare this to people who do not have HD. Because longitudinal studies often require large numbers of patients, the other goal for this study is to assemble a large database of patients who may be able to simultaneously participate in other clinical trials for HD. The Center has about 90 – 110 participants a year for this study.
Mr. Agrawal emphasizes that for observational longitudinal studies to get results, it takes time to be able to see statistically significant patterns that will further the understanding of HD. But these studies can provide critical information on the disease. For instance, the center has been able to demonstrate using sophisticated brain imaging, that striatal atrophy begins at least 10 years prior to clinical onset, that neuronal cell death correlates best with functional disability, and that the number of trinucleotide repeats in the huntingtin protein has an effect on the rate of HD progression. They found that individuals with the smallest number of trinucleotide repeats appear to have the best prognosis. For more about trinucleotide repeat lengths and the huntingtin protein, click here.
2) Genetic study for Huntington’s Disease-like 2 : Researchers at the Baltimore Huntington’s Disease Center are also very interested in looking for other genes that cause HD-like diseases. They have actually identified a number of these disorders, which are defined by having very similar symptoms to HD, but do not involve the altered HD gene- the HD gene can be perfectly normal and these symptoms may still appear. One of these diseases is called Huntington’s Disease-like 2 (HDL-2) and like HD, it is an adult onset neurodegeneration disorder, it is autosomal dominant, and it is characterized by chorea, cognitive, and psychiatric symptoms. It also involves neurodegeneration and inclusion bodies in the same parts of the brain as in HD. The Center is now working on a project to identify genetic markers for this disease, and to understand what kinds of mutations (other than the altered huntingtin mutation) could cause a disease like HD. They have recently published a paper in 2007, linking HDL2 to a glutamine/CTG repeat mutation on chromosome 16. This kind of mutation is related to, but not the same as the mutation involved in HD. They hope that by understanding the pathology behind HDL-2, it will shed new light on the pathology of HD and other neurodegenerative diseases.
3) Huntington Study Group trials : The BHDC participates in a number of multi-site clinical trials run by the Huntington Study Group. The Huntington Study Group is a non-profit organization composed of physicians, medical researchers and health-care providers from around the world. They have been organizing and conducting clinical trials for HD since 1993. In particular, the Center recruits participants for the observational PHAROS and COHORT studies, the PREDICT-HD study to find neurobiological markers of HD, and the completed TREND-HD study which is a therapeutic trial for Ethyl-EPA. For more on the Huntington Study Group, please click here, and for information on clinical trials in HD, please click here.
4) Memantine Study : The Center is also conducting a clinical trial with memantine, a drug that has already been approved by the FDA and marketed as Namenda for use in Alzheimer’s disease patients. However, not much is known about its use in HD, so the purpose of this clinical trial is to test if it will improve or delay cognitive symptoms of HD. The trial is sponsored by a company named Forest Pharmaceuticals, a company that produces drugs for cardiovascular diseases and CNS diseases like HD. This type of trial is a new indication study, and so it is run as a double-blinded, placebo trial over the course of 6 months. The Baltimore Huntington’s Disease Center started recruiting patients for this study in the fall of 2006, and as of March 2007 they have 72 participants. For more information on the use of memantine in HD, click here.
While anywhere from 150-200 people come in for clinical services in an average year, 2006 was an especially busy year for the clinic. Mr. Agrawal also explains that while many people come to the BHDC for genetic testing or patient care, about 25% to 50% of those who come also agree to participate in the clinical trials run through the Center. A good reason for any individual with HD to participate in the Longitudinal Core Study run at the Center is that there will be records of them kept in the Center’s database system, and so they can potentially be contacted to be recruited for any new therapeutic clinical trials if they fit the criteria needed to be included in the study.
Basic Research
Dr. Chris Ross is the director of the Baltimore Huntington’s Disease Center, and runs a research laboratory that focuses on the neuronal biology and genetics of neurodegenerative and psychiatric disorders such as HD, Parkinson’s disease, schizophrenia.
Dr. Ross discusses a few of the research projects conducted in his laboratory. He explains that the primary purpose of basic research is to identify biological targets for HD, and to develop those targets into treatments. For more on the process of going from basic research to a treatment, click here. As such, his lab focuses their research projects around potential biological targets for HD, and mostly on the huntingtin protein itself.
1) One area of current studies focus on how huntingtin aggregates are formed, and at what point these aggregates are toxic and cause nerve cell death. They have hypothesized that the larger aggregates or neuronal inclusions are not the most toxic molecules, but rather that the intermediates proteins in this aggregation pathway (the early aggregates) may be the most lethal form. As such, they may be the best molecules to target for future therapies. For more information on huntingtin aggregation, please click here.
Dr. Michelle Poirier, another faculty member at Psychiatry at the Johns Hopkins University School of Medicine, is doing studies in her own laboratory to investigate the shape or molecular structure of these intermediate huntingtin proteins. One of the most recent theories of how huntingtin protein aggregates are shaped describes them as made up of a series of folded strands of amino acids, with each strand composed of seven or eight glutamine amino acids. In a paper published in 2005, Dr. Poirier collaborated with the Ross lab to create two tissue culture models based on this theory. They confirmed that the proposed structure does indeed occur, and that there is a correlation between the presence of this type of huntingtin aggregate in the tissue culture cell and the presence of cell toxicity. However, they suggest that it is entirely possible that the toxicity is not caused directly by these aggregates, but rather that any of the kinds of intermediate species formed throughout the aggregate pathway may be responsible for toxicity instead.
2) Another project in the Ross lab looks at what kinds of proteins are involved in cutting the huntingtin protein into fragments. This process is also implicated in nerve cell toxicity in HD – it is thought that a fragment of huntingtin is actually more toxic to the cell than a full-length huntingtin protein. For more information on huntingtin protein fragments, please see figure P-2 here. In collaboration with Dr. David Borchelt and his laboratory at the University of Florida’s College of Medicine, Dr. Ross’ lab developed one of the initial transgenic mouse models of HD. Dr. Ross’s lab has used this mouse model to look at which enzymes play the largest role in generating toxic fragments of huntingtin protein. In collaboration with Dr. Michael Hayden’s laboratory they have already determined that caspase-6 is one of the most commonly involved enzymes in huntingtin fragmentation (please click here for a HOPES article on that finding), but they are looking at the role of other caspases and calpains (another family of proteases) as well.
Tamara Ratovitski, a member of the Ross lab, leads a related project using tissue culture models to look for the specific points in the chain of amino acids in the HD protein where fragmentation occurs. She wants to understand exactly how many fragments are generated by each type of protease, and how long they are. The goal is to target the most important proteases for inhibition, which will reduce the number of fragments and (presumably) cell toxicity.
3) A third project at the Ross lab looks at the affects of the mutant HD protein upon gene transcription. It is thought that the altered huntingtin protein changes the patterns of how genes are transcribed and translated, especially the genes that are key for a cell’s survival- and this may contribute greatly to toxicity in HD. Several years ago, Dr. Ross’ lab identified an unusual interaction between the mutant HD protein and the CREB-binding protein (also known as CBP), a smaller regulatory protein that is key for cell survival. For more information on the role of CBP in HD, click here.
Currently, the lab is following up this project with another group of studies intending to demonstrate a direct connection between the altered huntingtin protein, altered gene transcription, and cellular toxicity. It may be that the interaction between CBP and the altered huntingtin protein is one of a group of similar interactions between proteins involved in gene transcription and the altered huntingtin protein. If their research conclusively demonstrates that altered gene transcription does lead to cellular toxicity, one possible therapeutic intervention would be to use HDAC inhibitors. For more information on the potential role for HDAC inhibitors, please click here.
When asked what he thinks about the role of basic scientific research in respect to the larger body of HD research, Dr. Ross illustrates his opinion by discussing what goes on at the Baltimore Huntington’s Disease Center. Dr.Wenzhuan Duan, an assistant professor in the Department of Psychiatry and Behavioral Sciences works on research in Huntington’s and Parkinson’s disease. He takes biological targets identified through basic research on HD, and develops potential therapeutic drugs based on these findings. For more information on the process of drug research and development in HD, click here. They have developed a tissue culture model using nerve cells with the altered huntingtin protein that can be used to test potential therapeutic compounds to see if they might be useful for treating HD.
Dr. Ross emphasizes that another one of the major goals in doing therapeutic research is not only to cure HD, but to delay its onset. The idea would be to intervene by using treatments before the cognitive, motor, or behavioral symptoms actually appear. Members of the BHDC have already demonstrated that a great deal of neurodegeneration occurs before symptoms actually appear, so it would be effective if treatment occurred before the identifiable “onset” of symptoms to prevent or delay them. The Center has submitted a grant to conduct a phase II clinical trial to look at the effects of coenzyme-Q10 on presymptomatic HD patients, to see if it does in fact, delay the onset of symptoms. For more information of co-enzyme Q-10, please click here. The focus on research and treatments for presymptomatic HD patients is a very new direction for the Center, and appears to be a promising one.
For further reading
- The Baltimore Huntington’s Disease Center.
The website for this HDSA Center of Excellence.
- Rosenblatt A, et al. The association of CAG repeat length with clinical progression in Huntington disease. Neurology. 2006;66(7):1016-20.
This study demonstrates that individuals with the smallest number of trinucleotide repeats appear to have the best prognosis
- Reading S, et al. Functional Brain Changes in Presymptomatic Huntington’s Disease. Ann Neurology 2004;55;879-883
The 2004 publication demonstrating that there are significant structural changes in the brain in presymptomatic HD patients.
- Rudnicki DD, et al. Huntington’s Disease Like-2 Is Associated with CUG Repeat-Containing RNA Foci. Ann Neurology 2007;61;272-282
A follow-up study on HDL-2 demonstrating that it affects RNA function, and this may contribute to cell toxicity in HDL-2.
- Schilling G, et al. Characterization of Huntingtin Pathologic Fragments in Human Huntington Disease, Transgenic Mice, and Cell Models. J Neuropathology 2007. Vol 66, No. 4; 313-320
This publication demonstrates the location of significant sites of huntingtin fragmentation.
- Poirier MA, et al. A Structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet structure. Human Molecular Genetics, 2005. Vol. 14, No.6: 765-774.
This paper discusses the models for huntingtin protein aggregate structure, and further confirming the beta-strand/beta-turn model of aggregation.
- Wang W, et al. Compounds blocking mutant huntingtin toxicity identified using a Huntington’s disease neuronal cell model. Neurobiology of Disease. 2005;500-508.
This paper discusses a tissue culture model that has been demonstrated to be a good method to screen potential therapeutic compounds for treating HD.
-J. Seidenfeld, 8/21/2007
More
Dr. Rick Morimoto
Department of Biochemistry, Molecular Biology and Cell Biology
Northwestern University
Evanston, IL
Introduction
In November 2005, HOPES team member Justine Seidenfeld visited the Morimoto Laboratory in the Department of Biochemistry, Molecular Biology and Cell Biology at Northwestern University’s graduate school in Evanston, IL. HOPES would like to thank Dr. Morimoto and the members of his lab for taking time to meet with us and share their outlooks on research and Huntington’s Disease.
The Morimoto Lab studies how cells protect themselves by sensing and responding to environmental and physiological stress. These kinds of stress often lead to damage and problems within the cell. Stress can come from a wide variety of sources, like oxidative damage, over heating (heat shock), bacterial infections, and misfolded proteins (For more on proteins and their folding structure, click here) The cell has many different ways of recognizing and responding to stress induced problems. Under normal circumstances the cell can maintain a balanced state known as homeostasis. When stress causes proteins to misfold, the cell responds through a mechanism called the heat shock response. This response increases the amount of molecular chaperones and proteases produced within the cell. Molecular chaperones and proteases correct misfolded proteins, repair damage, and bring the cell back to balance. Sometimes, however, there are so many misfolded proteins that the heat shock response cannot keep up. The misfolded proteins accumulate into protein aggregates. Aggregates are a hallmark of neurodegenerative diseases like HD.
Approaching HD
The Morimoto Lab studies the cell’s response to the stress-related buildup of misfolded proteins within the cell. These researchers are attempting to understand the mechanisms that lead to the appearance of misfolded proteins in the cell in the first place. They also study how these misfolded proteins affect the heat shock response, and the consequences for the cell’s overall health.
The Morimoto Lab approaches these questions from a variety of angles. They have pioneered the use of a roundworm, or C. elegans, model to study HD. They created a mutant of C. elegans which carries the gene for an expanded polyglutamine chain protein that is characteristic of HD. Furthermore, they have shown that the polyglutamine chain by itself is toxic, not only when it is part of an altered huntingtin protein. (For more on the structure of the altered huntingtin protein, click here) They have also conducted a genetic screen of the entire genome of these nematodes to determine what other proteins interact with the polyglutamine chain protein, and find out how to suppress polyglutamine protein aggregation. Among the 186 proteins identified, many are molecular chaperones involved in the heat shock response. So in order to understand exactly how altered huntingin protein aggregation occurs in the cell, the lab is studying the interaction between molecular chaperones like Hsp70, its co-chaperone Hsp40, and aggregates .
Research
Dr. Morimoto’s lab looks at HD from the very beginning of the disease cascade. He describes the disease cascade process like this: a protein with an expanded polyglutamine chain (which is toxic) impairs cells and their function over time, eventually leading to nerve cell death. His lab studies the beginning of the cascade, asking the question “What goes wrong in protein quality control?” Dr. Morimoto believes that protein quality control is one of the cell’s most ancient mechanisms. In the very first forms of life, RNA was the main hereditary genetic material in cells, instead of DNA as it is now. In this “RNA world,” cells were much less complex. In order to survive all of the sources of stress they were subjected to, they must have had very good protein quality control mechanisms. These mechanisms have been preserved in the “DNA world” of the present day. Additionally, the question of protein quality control is especially intriguing because protein misfolding is common among neurodegenerative diseases, HD included. Dr. Morimoto’s findings have the potential to be useful in the study of other neurodegenerative diseases. “The implications are broad,” he says of his research program.
Why does Dr. Morimoto focus on HD if is research is so broad? As he puts it, “HD is a leader as a genetic dominant.” As opposed to some other neurodegenerative diseases, HD is a disease that is easy to identify by using genetics. The presence of at least one allele for the altered Huntingtin gene always indicates a person will have HD. The genetic link is not so clear in other neurodegenerative diseases. For example, only 2% of cases of amyotrophic lateral sclerosis are actually related to an inherited genetic mutation. These cases are called Familial Amyotrophic Lateral Sclerosis, or FALS. In short, using HD to study protein quality control is valuable because it has straightforward genetic links, and it has broader implications for other neurodegenerative diseases.
The Morimoto Lab has also created C. elegans models related to tau pathologies, amyotrophic lateral sclerosis, and other polyglutamine diseases besides HD. All of these models help to understand the genetic basis of protein misfolding by comparing the different molecules associated with each of these diseases. In addition to C. elegans, the lab uses use in vitro studies and tissue culture (cells grown in a Petri dish), among others as model systems to answer these questions. Dr. Morimoto describes these multiple approaches as “the only way to run a lab.” These different models allow the lab to run multiple tests on hypotheses, so they can be much more confident about their results. For example, Dr. Morimoto says that there are certain advantages to doing experiments with tissue culture, but that they need to be verified in studies using whole organisms. In doing this, they can see if they get the same in an entire living animal, not just isolated tissue in a Petri dish, and the C. elegans model is a good way to test any hypothesis in his lab.
Dr. Morimoto also discusses the pharmacological studies done in his lab. In these studies, the researchers discover small molecules involved in protein folding. These molecules could serve as a basis for therapeutic drugs or therapy treatments. In a large collaborative effort with 26 other laboratories worldwide, the Morimoto lab helped identify a small molecule called celastrol, from a plant often used for its anti-inflammatory properties in Chinese herbal medicine. Celastrol was identified because it can activate important genes involved in the heat shock response and elevate the level of molecular chaperones in the cell. These molecular chaperones could help protect the cell from the damage caused by misfolded proteins. Members of the Morimoto lab are now working with Dr. Richard Silverman, a professor in the Department of Chemistry, to discover the specific structure of celastrol and how it works with other proteins to activate the heat shock response. Dr. Morimoto believes a detailed understanding of exactly how celastrol works is important before it is turned into a therapy drug and put into clinical trials. However, the potential of celastrol certainly seems promising.
HD Research and the Public
Dr. Morimoto discussed his views on the responsibilities of a scientist to the public. “It is our responsibility and obligation to talk to the public about research,” he says. Dr. Morimoto wants patients and their families to understand how research on neurodegenerative disorders works: how scientists pose questions; how drugs and therapies are created; and why animal models like flies, worms, and mice, are used instead of humans at certain stages of the research process.
Dr. Morimoto explains that using a model system like C. elegans is helpful because this organism has a lot of important similarities to humans, but also differences that make them easier to study. C. elegans has a much simpler nervous system than humans. C. elegans has only 302 nerve cells, as compared to the human nervous system which can have 10 to 100 billion nerve cells. Although the C. elegans nervous system is less complex than ours, each individual nerve cell (as opposed to the whole system) is just as complex as a human’s nerve cell. C. elegans is good in vivo model for the Morimoto Lab, because they are focusing on protein balance and health within the individual nerve cell rather than an entire nervous system.
Dr. Morimoto believes that he and other academic scientists are fortunate because they can choose to focus their research on whatever interests them. They can be well-funded, but that gives them an obligation to educate people on how their money is being spent. Yet not all scientists are trained to do this. Dr. Morimoto believes that more effort is needed to increase communication between laboratory researchers and the public.
Dr. Morimoto is one of the 17 Huntington’s Disease Society of America (HDSA) Coalition for the Cure Senior Researchers. He is committed to long-term HD research and collaborates with other scientists around the world who also study HD. Through the local chapter of the HDSA, HD patients and their families often contact him, and he will sometimes bring them to his lab to meet the postdoctoral fellows, graduate, and undergraduate students working to discover the mechanisms behind HD. Dr. Morimoto says that it helps him get a sense of clarity apart from his research, and he thinks that it is equally beneficial to the patients and their families. He also attends Illinois HDSA meetings to give talks. Since he knows that patients and families are always at these meetings, he tries to make his talks especially relevant for them. One of the great things about science, he says, is that all scientific information is accessible. And the local chapters of the HDSA get scientists to appreciate the public, to make sure that information reaches them in an appropriate way. To learn more about the HDSA, click here.
A unique feature of HD is that it is relatively rare compared to some other neurodegenerative disorders like Alzheimer’s and Parkinson’s (for more information on these diseases, click here). Because less people are affected by HD, there is less funding from the National Institutes of Health (NIH) devoted to HD. Dr. Morimoto says that the community of HD research advocates is amazingly strong and plays a big role is raising necessary money. As an example, he cites the Fiore family of Highwood, IL. The Fiores have a history of HD in their family, and have raised money for HD research by partnering with the HDSA and holding their own fundraising events like golf and poker tournaments (you can visit their site by clicking here). Their work has helped to raise money to open an HDSA Center of Excellence at Rush St. Luke Medical Center in Chicago. According to Dr. Morimoto, families like the Fiores have single-handedly used their personal adversity to “increase awareness of the disease for all.”
More on Dr. Morimoto
Dr. Morimoto received his Ph.D. in molecular biology from the University of Chicago in 1978. He says that he became interested in his line of research when, as a graduate student, he heard a talk by Dr. Matthew Meselson of Harvard University. Dr. Meselson spoke about the newly discovered heat-shock response system in fruit flies. Dr. Morimoto was intrigued by this talk, and did his postdoctoral fellowship at Harvard in Dr. Meselson’s lab. While he was there, he was the first to clone the human Heat-Shock Protein 70 (Hsp70) in 1985, This means that he was the first to discover the location and sequence of the gene that codes for the protein. Now it could be used in genetic studies related to the heat shock response. Hsp70 was identified as a key protein in protein quality control. Multiple studies showed that by making an unusually high amount of Hsp70 in cells prone to protein misfolding, it would suppress the toxic effects of these misfolded proteins. Dr. Morimoto came to Northwestern University in 1982 and continued his research on the heat shock response in neurodegenerative diseases. As he puts it, the 1970s and 80s were full of important discoveries about the regulation of protein folding related to the heat-shock response. The 90s brought increasing awareness that neurodegenerative diseases were related to the misfolding of protein aggregates. Seven years ago, his lab began to use C. elegans to study the heat shock response. They began to also study HD in C. elegans, and this is a major portion of his current research. Dr. Morimoto says that his interest in science stemmed from, and continues to be, a desire for pure knowledge, to understand the natural world.
Future of HD Research
What are some of the greatest obstacles facing the community of researchers studying on HD? Dr. Morimoto believes that one of the most pressing questions involves the use of model systems like C. elegans, mice, or tissue culture to study HD. We can certainty understand the fundamentals of protein folding, aggregation, toxicity, and nerve cell death by using these model systems. However, to what extent can we take these fundamental discoveries and apply them to humans- a vastly more complex creature than mice, worms, or yeast? Dr. Morimoto believes that discoveries in model systems will prove to be useful and important because the machinery behind protein quality control is an ancient and conserved process. He believes that most of the discoveries made in less complex model systems will probably also be found in humans. However, he is concerned that researchers will have to take into account the fact that humans have very complex systems (such as human metabolism) when developing therapies. Another obstacle will be that of developing effective drug therapies. He explains that there has been limited success in discovering which proteins to target with therapeutic drugs . One in one hundred options may work, but there are not yet one hundred likely targets. In order to discover new targets, Dr. Morimoto argues that there must be more consensus among different branches of the scientific community. He strongly believes that when research laboratories partner up, they are much more effective and successful than when they work on their own.
Dr. Morimoto emphasizes that, among the myths of research that he would like to dispel, research labs like his are wonderfully diverse in their scope of cultural backgrounds, scientific experience, and intellectual perspectives. He stresses that the lab environment can be a very cooperative and social one, as opposed to the stereotype of fierce competition. Diversity and collaboration are important characteristics for successful scientific research.
-J. Seidenfeld, 3/14/06
More
This chapter explains what it means to be an HDSA “Center of Excellence”. We begin with a brief description of HDSA, the organization that names Centers of Excellence. Next we describe many of the services that the Centers of Excellence offer. We include a description of what an HD patient could expect during a first visit to a Center of Excellence clinic. And finally, we give a full list of the HDSA Centers, with contact information for each.
The Huntington’s Disease Society of America
There are nineteen HDSA Centers of Excellence located in the United States.
All of these Centers have specialized HD programs and reputations for first-rate treatment. But how are they chosen and what exactly is required to become a Center of Excellence?
Centers of Excellence are chosen by the Huntington’s Disease Society of America (HDSA). HDSA is a national nonprofit health organization dedicated to finding a cure for Huntington’s Disease and to providing support for everyone affected by HD – both patients and their families. For more information about HDSA, or to contact HDSA, click here.
By recognizing some of the best HD clinics in the nation, HDSA hopes to promote the highest standards of care. To qualify as a Center of Excellence, an HD clinic must provide comprehensive services, have a reputation for excellent medical care, and demonstrate a commitment to HD education and research.
What it means to be an HDSA Center of Excellence
All Centers of Excellence maintain an active HD clinic. The clinic not only serves patients already diagnosed with HD, but is also equipped to provide pretest counseling and genetic testing. Centers of Excellence typically have a staff of specialists who can help with issues specific to HD. For example, many Centers have physical therapists, neurologists, genetic counselors and speech therapists. This specialized staff is not the only reason why Centers of Excellence are especially qualified to treat HD. Everyone working at the Center also has a familiarity with the disease. The Staff at Centers of Excellence know what individuals with HD and their families need based on extensive experience. The Centers also always have the most recent and accurate information available.
Additionally, every Center of Excellence can offer patients the opportunity to participate in clinical trials and other HD research opportunities. Many centers are part of the Huntington’s Study Group (HSG). HSG is a nonprofit group of healthcare providers based in the United States, Europe, Canada, and Australia who join together to conduct clinical research trials. By organizing and coordinating research across large numbers of sites, HSG can conduct comprehensive clinical trials. To visit the HSG website, click here. Participating in clinical trials can be a way to contribute to the HD community and become part of the effort to improve the lives of those affected by the disease.
All HDSA Centers of Excellence engage in educational outreach. The centers have several ways of providing information about the disease. Centers will usually have comprehensive literature available at the clinic for patients. This can be especially helpful for individuals who have recently discovered they have HD. Additionally, some centers hold conferences where many groups and individuals can gather and exchange information. These conferences let researchers, doctors, organizations and others committed to treating and caring for HD patients stay on the forefront of the latest research findings. Finally, many Centers attempt to share their expertise with other medical workers caring for HD patients. For instance, the Center can send staff directly to local nursing homes to provide HD specific training, information, and assistance to the nursing home staff. This outreach is an important aspect of the Centers’ mission to provide care and service to the HD community.
The Role of the HD Clinic
Many newly diagnosed HD patients do not know what to expect from an HD clinic. The thought of going to a clinic specifically focused on HD can seem intimidating and daunting. The goal of this section is to give a brief description of what happens during a first time visit to an HD clinic. Hopefully, this knowledge will help make the experience easier and more comfortable for everyone involved.
In many ways, going to an HD clinic is much the same as a normal doctor’s visit. The main difference is that the clinic staff members are guaranteed to have specialized knowledge about HD. Although the staff will always tailor treatment to the individual patient, they have a broad base of experience to draw upon; they understand what it means to have HD and to be at risk for HD. Additionally, the clinic may have resources helpful to HD patients that a regular doctor wouldn’t have (such as HD-experienced physical therapists, social workers, genetic counselors, etc.).
During a first visit the patient will always see a doctor. They may or may not see other HD specialists depending on individual circumstances. Most of the time they will see other specialists during follow-up visits. The length of time between visits varies dramatically depending on individual circumstances. If the doctor decides that only a routine follow-up visit is required, the next visit may be scheduled for one year later. If the doctor has a specific issue he or she wants to address, a follow-up visit may be scheduled as soon as one month later. There is no standard length between visits – it is entirely dependent on the patient’s needs and desires.
Finally, HD clinics at Centers of Excellence are active in current clinical trials. This is one of the requirements a clinic must meet to be chosen as an HDSA Center of Excellence. Depending on the status of ongoing trials, the clinic could offer patients the opportunity to participate. These clinical trials provide a chance to contribute to HD research. The exact nature of the available trials will vary from clinic to clinic and change over time.
We hope patients, or potential patients, will not feel intimidated by the idea of visiting an HD clinic. The clinics exist to help and support the patients and their families. Clinic doctors and personnel understand the difficulties of living with HD and their goal is to offer the best care available. We suggest that everyone in the US with HD and their families consider a visit soon to an HD Center of Excellence, if they haven’t already.
List of HDSA Centers of Excellence
For an interactive map, listing HDSA centers of excellence by state, click here.
Northeast
New England HDSA Center of Excellence
Charlestown , MA 02129
Director: Steven Hersch, M.D.
Center Contact: Jamie Hill
Phone: 617/724-2227
Email: jhill11@partners.org
Website: http://www.mgh.harvard.edu/neurology/services/treatmentprograms.aspx?id=1048
HDSA Center of Excellence at the University of Rochester
Rochester , NY 14620
Directors: Kevin Biglan, M.D., MPH and Frederick Marshall, M.D.
Center Contact: Leslie Briner
Phone: 585/273-4147
Email: leslie.briner@ctcc.rochester.edu
HDSA Center of Excellence at Columbia Health Sciences/NYS Psychiatric Institute
New York , NY 10032
Directors: Karen Mardner, M.D.
Center Contact: Debbie Thorne
Phone: 212/305-9172
Email: thorned@sergievsky.cpms.columbia.edu
Website: http://www.hdny.org/
George G. Powell HDSA Center of Excellence at North Shore University Hospital
Manhasset , NY 11030
Director: Andrew Feigin, M.D.
Center Contact: Patricia Lambert
Phone: 631/234-0136
HDSA Center of Excellence at Johns Hopkins University/Johns Hopkins Hospital
Baltimore , MD 21287
Directors: Christopher Ross, M.D., Ph.D. and Adam Rosenblatt, M.D.
Center Contact: Debbie Pollard
Phone: 410/955-2398
Email: dpollard@jhmi.edu
HDSA Center of Excellence at the University of Virginia
Charlottesville , VA 22903
Director: Madaline Harrison, M.D.
Center Contact: Pat Allinson, M.S.
Phone: 434/924-2665
Email: psa9m@hscmail.mcc.virginia.edu
Southeast
HDSA Center of Excellence at Emory School of Medicine
Atlanta , Ga. 30329
Directors: Randi Jones, Ph.D. and Claudia Testa, M.D., Ph.D.
Center Contact: Joan Harrison, G.N.P.
Phone: 404/728-6364
Email: jharri2@emory.edu
HDSA Center of Excellence at University of Alabama
Birmingham , Ala. 35294
Director: Leon Dure, M.D.
Center Contact: Stacey Mantooth
Phone: 205/996-7850
Email: smantooth@peds.uab.edu
HDSA Center of Excellence at the Univeristy of South Florida Huntington’s Disease Clinic
Tampa, FL 33612
Director: Juan Sanchez-Ramos
Center Contact: Kelly Elliott, RN
Phone: 813/974-6022
Email: kelliot@health.usf.edu
Midwest
HDSA Center of Excellence at Ohio State University
Columbus , Ohio 43210
Director: Sandra Kostyk, M.D., Ph.D.
Center Contact: Nonna Stepanov
Phone: 614/688-8672
Email: stepanov-1@medctr.osu.edu
HDSA Center of Excellence at University of Iowa Hospitals and Clinics
Iowa City , Iowa 52242
Directors: Jane Paulsen, Ph.D. and Robert Rodnitsky, M.D.
Center Contact: Anne Leserman
Phone: 319/353-4307
Email: anne-leserman@uiowa.edu
HDSA Center of Excellence at Hennepin County Medical Center
Minneapolis , MN 55415
Director: Martha Nance, M.D.
Center Contact: Dawn Radtke, RN
Phone: 612/873-2943
Email: Dawn.radtke@yahoo.com
HDSA Center of Excellence at Washington University School of Medicine
St. Louis , MO 63110
Director: Joel Perlmutter, M.D.
Center Contact: Stacey Barton, MSW, LCSW
Phone: 314/362-3471
Email: bartons@neuro.wustl.edu
HDSA Center of Excellence at Rush University Medical Center
Chicago , IL 60612
Director: Kathleen M Shannon, M.D.
Contact: Jean Jaglin, RN, CRC
Phone: 312/942-5003
Email: Jean_A_Jaglin@rush.edu
HDSA Center of Excellence at Indiana University
Indianapolis, IN 46202
Directors: Kimberly A Quaid, Ph.D and Joanne Wojcieszek, M.D.
Contact: Leo Rafail, BSW
Phone: 866/488-0008
Email: info@iuhdclinic.org
West and Pacific Northwest
HDSA Center of Excellence at Colorado Neurological Institute
Englewood, CO 80113
Director: Rajeev Kumar, M.D.
Center Contact: Mimi Ortiz
Phone: 303/357-5453
Email: ortiz@kumarneuro.com
HDSA Center of Excellence at University of Washington
Seattle , Wash. 98195
Director: Thomas Bird, M.D.
Center Contact: Debbie Olsen (general info)
Phone: 206/616-2135
Southwest
HDSA Center of Excellence at University of California Davis Medical Center
Davis Medical Center
Sacramento , Calif. 95817
Directors: Vicki Wheelock, M.D.
Center Contact: Teresa Tempkin R.N.C., M.R.N., A.N.P.
Phone: 916/734-6278
Email: teresa.tempkin@ucdmc.ucdavis.edu
HDSA Center of Excellence at University of California, San Diego
San Diego , Calif. 92103
Directors: Jody Corey-Bloom, M.D., Ph.D.
Center Contact: Jody Goldstein
Phone: 858/622-5854
Email: jlgoldstein@ucsd.edu
HDSA Center of Excellence at University of California, Los Angeles
Los Angeles , Ca 90095
Director: Susan L Periman, M.D.
Center Contact: Sakena Patterson
Phone: 310/794-1225
Email: sjpatterson@mednet.ucla.edu
HDSA Center of Excellence at Baylor College of Medicine
Houston , Texas 77030
Director: Joseph Jankovic, M.D.
Center Contact: Alicia Palao
Phone: 713/798-7438
Email: palao@bcm.edu
For Further Reading
- http://www.hdsa.org– The official website of the Huntington’s Disease Society for America.
- http://www.huntington-study-group.org/ – The official website for the Huntington Study Group. This site contains information concerning ongoing clinical trials, as well as brief summaries of previously completed trials.
-A. Hepworth, 1-24-07, updated 6-21-11
More
Dr. Diana Rosas
Massachusetts General Hospital
Charlestown, MA
Dr. Rosas is a clinician who treats people with HD. The complexity and mysterious nature of the illness inspired her to get involved with Dr. Nancy Wexler’s Venezuela project in the early 1980s—a genetic study of large families that were all descended from a single ancestor with HD. The goal of the project was to generate a precise map of genetic markers.
Since then, Rosas has dedicated her life to treating the symptoms experienced by people with HD, as well as to investigating the effects of HD on the brain. In contrast to researchers like Drs. James Gusella and Marcy MacDonald (please see Chapter 1, Part 1 of Research Frontiers who direct their efforts toward the root causes of HD and the beginning of the disease cascade, Rosas and other clinicians focus on the end of the cascade, in which motor, cognitive, and psychiatric functions begin to decline. Rosas defines the age of onset of HD as the age when motor symptoms (irregular eye movements, chorea, etc.) appear. Interestingly, some people are able to suppress their chorea during a doctor’s examination, and, as a result, their HD goes undiagnosed for a long period of time.
Rosas also discussed the unique onset of juvenile HD. Children with greater than 45 CAG codon repeats in the Huntington gene have an earlier and much more severe onset than people with traditional HD. While these children experience less chorea than their adult counterparts, they often have hallucinations and symptoms that resemble those of Parkinson’s disease.
Although she acknowledges that it is important for clinicians to concentrate on the striatum, where neurodegeneration starts, Rosas mostly takes a holistic approach to the brain. She emphasizes that the entire brain, not just the striatum, deteriorates as a result of HD.
“The motor cortex is affected across the board,” said Rosas. Located on the frontal lobe, the motor cortex carries out the initial processing of motor information in the brain and is involved in the control of movement.
The earliest changes in the brain occur in the posterior regions (the parietal, occipital, and temporal lobes). During the course of the disease, however, a person experiences large reductions in volume in almost all areas of the brain. For example:
To learn more about the different parts of the brain, please click here.
The treatment of HD requires an integrated, multidisciplinary approach that includes the medical management of symptoms; physical, occupational, and/or speech therapy; and genetic and psychological counseling. In addition to treating people who already have the disease, Rosas counsels people who are at risk for developing it. While the decision to undergo genetic testing is a very personal one, Rosas does recommend it for those who are planning to have children.
She encourages all others at risk not to undergo testing for several reasons. First, there is no cure for HD. Second, insurance issues and genetic discrimination in the workplace may result if an at-risk person finds out that he or she is likely to get the disease. Third, knowing that one is likely to get HD would cause unnecessary worry. He or she would constantly wonder, “When will I get sick?” Fourth, even negative test results may cause emotional distress, such as “survivor’s guilt” if one’s parents died from HD.
Finally, Rosas strongly advocates seeing a doctor for a complete neurological examination as soon as one notices the first physical symptoms of HD. She advises people not to wait. A doctor is the best judge of whether or not someone has the disease. Sometimes, though, people do experience sexual, mood, and marital problems even before the onset of motor symptoms. These problems are symptoms associated with the psychosocial part of the disease, and are most common in men. Many psychosocial symptoms can be treated by clinicians and psychologists.
For those who have been diagnosed with HD and are interested in participating in clinical trials, Rosas recommends visiting the website of the Huntington Study Group. Among its many resources, the website provides information about clinical trials in progress and new clinical trial initiatives.
-T. Altman, 5/5/06
More
Drs. James Gusella and Marcy MacDonald
Molecular Neurogenetics Unit, Massachusetts General Hospital
Charlestown, MA
Introduction
This summer two HOPES team members, Taylor Altman and Shawn Fu, spent several days with Drs. James Gusella and Marcy MacDonald at their laboratories in the Molecular Neurogenetics Unit of Massachusetts General Hospital (MGH) in the Boston area. HOPES would like to thank Gusella and MacDonald for taking the time to share many valuable insights with us.
The Molecular Neurogenetics Unit, under the direction of Gusella, is dedicated to using molecular genetic techniques in humans and mice to understand and treat neurological disorders. Gusella and his colleagues pioneered the use of DNA sequence polymorphisms as genetic markers and mapped the Huntington gene to chromosome 4 in 1983 (please click here for more information about this breakthrough). He and his fellow researchers have since applied this genetic mapping approach to numerous neurological diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), Batten disease, and familial dysautonomia (FD) in an effort to pinpoint the chromosomal locations of the disease genes and/or the nature of the genetic defect itself. In 1993, Gusella, MacDonald, and their colleagues, as part of an international collaboration, identified an expanded, unstable CAG trinucleotide repeat in a novel gene as the cause of Huntington’s disease.
Both Gusella and MacDonald are currently trying to understand the pathogenesis of HD and find ways to interfere with the disease cascade in order to halt its progression. According to the webpage of the MacDonald laboratory, she and her colleagues follow three interrelated steps in order to investigate the complex biology behind HD and other neurological disorders:
- The use of polymorphic DNA markers in genetic family studies to isolate gene defects that cause inherited disorders of the nervous system and test loci that alter the disease phenotype in some way.
- The creation of model systems (such as the mouse model of HD) in order to understand and clarify the functions of the products (essentially, proteins) of the abnormal gene at the whole animal, cellular, and biochemical levels.
- The use of model systems to explore novel gene therapy based strategies, which aim to intervene in the disease process.
MacDonald uses knock-in mice that carry human mutations, such as the gene defects in HD and Batten disease, to identify early cellular and molecular events in the disease pathways. She is also in the process of identifying cellular partners (the parts of the cell that a chemical or molecule interacts with) and pathways for the HD and Batten disease proteins, huntingtin and battenin.

The MacDonald lab
What’s special about HD research?
Altman and Fu sat down with Gusella and MacDonald to discuss their passion for investigating HD. What’s special about HD research? Both stated that they have the opportunity to do a great service for those with HD and, at the same time, learn about the biology behind the disease.
“HD, in particular, gets scientists interested,” said MacDonald. “What makes [HD research] fun is the discovery part, being in the middle of nowhere and finding new biology.” Take, for example, the mutant huntingtin protein: “We have no clue what it does, but we can connect it with old things and see things that are brand-new.” When studying the biology behind non-genetic (traditional) disease, researchers learn from what they already know, which is “not great for discovery,” according to MacDonald. Conversely, when studying HD, researchers do not know what they are looking for, which sets the stage for some amazing new findings.
“What’s disturbing is that the more you know about how something works, the less interesting it is,” Gusella said, pointing out a negative aspect of scientific discovery. “But working on HD is different”, MacDonald asserted. Unlike other degenerative disorders of the nervous system like Alzheimer’s and Parkinson’s disease, whose “ etiologies are myriad” (meaning that there is a wide range of possible causes), everyone who develops HD has the same mutation. The hypothesis that things will be found that counteract the effects of the single mutation in the single gene that triggers the disease in everyone is exciting to work on because it still poses new and interesting questions and challenges for the scientists who work on it. (For a comparison of Alzheimer’s, Parkinson’s, and HD, please click here.)
Although MacDonald, Gusella, and their colleagues are interested in many aspects of the Huntington gene and related proteins, the overarching purpose of their work is finding a way to treat HD sufferers. “The ultimate goal of our research is a treatment, not understanding HD completely,” said Gusella. MacDonald added, “If you know how the disease starts, there will be a solution.” However, Gusella believes that an actual cure will not be possible until the HD allele vanishes from the population – something that is not likely anytime in the foreseeable future (he defines “cure” as the disappearance of the HD allele from the gene pool). (For more information on the distinction between a treatment and a cure, please click here.)
Conceptualizing HD
MacDonald drew a diagram of the disease (please see Figure AH-2: “HD pathogenesis and progression” below), its original CAG repeat mutation, and the age of onset. She and Gusella are currently looking for genes that interact with the original mutation. They conceptualize the disease in the human body as a series of steps in time from the HD CAG mutation to the onset of nerve cell death, to clinical symptoms and then the progression of the symptoms with factors that can effect each step (modifiers) determining the rapidity of each step and the age of onset of symptoms. Their labs are working to produce information about the genetic factors that determine the age of onset in the HD-MAPS (HD Modifiers in Age of Onset in Pairs of Siblings) study, coordinated by Dr. Richard H. Myers of the Boston University School of Medicine. By understanding the biological modifiers of onset, researchers may be able to develop methods to delay the onset of HD. Similarly, in the Huntington’s Disease PREDICT-HD (Neurobiological Predictors of Huntington’s Disease) study, conducted by Dr. Jane S. Paulsen of the University of Iowa, individuals who are known to have the gene expansion for HD are continually being recruited for a brief study examining changes in the DNA (genetic factors) that influence the age at onset of the disease.

Figure AH-2: A conceptual diagram of HD
Approaching HD
How do scientists go about studying HD? Gusella and MacDonald explained the approach they take to their research. They look at HD from a genetic angle, choosing to study the basics – in other words, the root cause(s) of the disease – not just the symptoms and pathology (the clinical aspect that interests doctors). If we envision the disease cascade as a timeline, Gusella and MacDonald work at the very beginning, understanding the various mechanisms that trigger the disease, including the early biochemical and metabolic changes that take place. Clinicians, on the other hand, work near the end of the cascade, looking not at the cause(s) of the disease, but rather at the consequences (the movement, cognitive, and psychiatric symptoms). Whereas clinicians generally try to intervene toward the end of the disease cascade with drugs or other therapies, Gusella and MacDonald attempt research that aims to intervene at the start of the cascade via experimental methods on the level of RNA, most notably RNA interference (RNAi), which will be discussed later. The researchers even anticipate that someday they may be able to change the slope of the cascade so that the age of onset is later and the symptoms are less severe. (As you may already know, the later the age of onset, the less severe the symptoms.)
Genetic disease is fundamentally different than non-genetic disease in that it has a definite starting point, even if symptoms are not yet apparent. In contrast, non-genetic disease usually has apparent symptoms that clinicians try to block, but not always an easily identifiable starting point. In the study of HD, researchers like Gusella and MacDonald have the broadest definition for the “disease” part of Huntington’s disease. They define it as the genetic mutation present at the time of conception all the way to the last stages of the cascade near the end of the person’s life. Neuropathologists (professionals who study diseases of the nervous system) define the disease as nerve cell death. Clinicians, who have the narrowest definition, consider it to be the visible symptoms in the last stages of the cascade.
MacDonald argues that huntingtin protein aggregation and eventual nerve cell death are not the causes of the disease, but rather the consequences. She and Gusella, therefore, strive to discover the things that happen before, without worrying about integrating the results of their experiments into the HD “story.” For a decade, the HD story has sounded the theme of huntingtin aggregation as a cause of HD. Here the term “story” describes a paradigm created by the scientific community – something of an umbrella idea under which many subsequent research findings huddle. While a story of this kind may one day be proven false, the scientific community has a hard time letting go or changing the paradigm. Similar to the HD story, the Alzheimer’s story emphasizes the buildup of amyloid plaques in the brain as the cause of the disease (the events that start the disease cascade). This story, too, may turn out to be inaccurate. Protein clusters may not cause either disease, but are considered to be the result of events that occur further upstream in the disease cascade. In other words, once the original trigger (as yet unidentified) sets off the disease cascade, it often takes a long time for protein clumps or tangles to form.
To hypothesize or not to hypothesize?
“The first step [of experimentation] is observation, not a hypothesis”, Gusella said. The scientific community has been so focused on having a hypothesis, he said, that getting funding without one was almost impossible because research projects that appeared purely descriptive (that is, projects that seemed to merely describe a phenomenon rather than answer a research question) were rarely awarded grants in the recent past. The scientific establishment’s penchant for storytelling may still prompt some researchers to formulate questions that they already know the answers to (“I can map and clone this gene”, for instance) simply to get funding for experimental research that may inhibit the careful observation necessary to formulate a meaningful hypothesis.
“Unfortunately”, said Gusella, “scientists must tailor their results to fit a hypothesis, which then must fit the overarching story that the research community is trying to tell.” This need for storytelling often results in unnecessary hype over a “rediscovered” discovery. For example, Alzheimer’s researchers have presented the same or similar findings about amyloid plaques over and over again, and the scientific community treats these findings as new discoveries. The way MacDonald views it, “It’s like mentioning Wilt Chamberlain to today’s great basketball players, and they say, ’Who?’”
Gusella and MacDonald think of science as a field in which researchers must continually build on the past, or else they run the risk of repeating the past (as in the Alzheimer’s example) or even losing it (as in the Wilt Chamberlain example). Gusella, though, cited an example of a scientific endeavor that did not have a hypothesis but was ultimately successful – the Human Genome Project, a thirteen-year effort coordinated by the U.S. Department of Energy and the National Institutes of Health to determine the complete sequence of the DNA in the human genome. The project gathered genetic information and developed a hypothesis later. Thankfully, said Gusella, the scientific community is beginning to come around; nowadays, more value is placed on descriptive research.
“Regardless of the type of research one is doing,” Gusella concluded, “the results can be interpreted in various ways, but one must select the correct interpretation.” Scientists must be willing to rule out certain hypotheses, as well as present failed hypotheses to their peers and to the public. Negative results are just as important as positive results because the data from a failed experiment may be useful in future research.
Accomplishments
Gusella, MacDonald, and their colleagues in the field of HD research have accomplished much in their careers. As mentioned earlier, Gusella mapped the Huntington gene to chromosome 4 in 1983. Dr. Gillian Bates of King’s College, London, cloned the tip of the chromosome, where the Huntington gene is located, and with Gusella and MacDonald, made a road map that pointed to the location of the gene. From there, researcher Christine Ambrose narrowed down the search for the gene to the two most promising segments of the tip, IT15 and IT16 (IT is lab lingo for “Interesting Transcript”). She discovered that these ITs were actually one gene, the Huntington gene. (Several neighboring ITs are now known to be inherited together with this gene.) Then, with her colleague Mabel Duyao, Ambrose made an assay that found that excess CAG repeats on the HD allele of the Huntington gene cause HD.
MacDonald got involved in the research effort in 1985, discovering new pieces of DNA on chromosome 4. In 1987, enough pieces of the chromosome were cloned to show various gene recombination events. Recombination events occur when genes lying farther apart on the same chromosome are often not inherited together due to the transfer of segments of DNA between homologous chromosomes during meiosis, the reproductive process by which sperm and egg cells are made. A haplotype study (a study describing the genetic makeup of an individual with respect to a specific pair of alleles or genes) was published in 1992. The objective of the study was to find out if the chromosomes on which the Huntington gene resides looked similar at a particular region in different people who had HD (in other words, the researchers wanted to know if the same mutation causes HD in everyone who has it). Through linkage studies (for more information, please click here), the researchers found that there was more than one mutation in ancestral populations (the predecessors of various groups of people who suffer from HD today). Interestingly, they also found the same mutation at the same spot in chromosome 4 in unrelated people with HD, so the same type of mutation in the same gene causes the disease in all people.
MacDonald’s Masterminds
Altman and Fu were fortunate enough to get a behind-the-scenes look at where HD breakthroughs are being made. They attended a lab meeting, where several of MacDonald’s postdoctoral (post-PhD) research fellows and interns gave presentations about their individual research projects. “Presenting research and sharing results is an important part of science,” said MacDonald.
Research fellow Julie Woda discussed her work on huntingtin normal function. Past research has shown that the protein is quite large and contains 36 HEAT-like repeats. The acronym HEAT comes from four proteins in which these repeated sequences have been found. These HEAT repeat sequences, which fold up into a spiral structure (alpha helix-loop-alpha helix), may serve as docking sites for other proteins. Most huntingtin partner proteins (“binding partners”) bind to one of its end segments. Based on its binding partners, huntingtin is involved in a variety of processes in the cell, including signal transduction, regulation of transcription (a big step in the process of turning DNA code into a protein), trafficking of molecules and other materials within the cell, maintaining the function of the cell’s cytoskeleton, and gene splicing.
Woda believes that the normal huntingtin protein is essential for the development of the human embryo. Studies conducted in the past have revealed that inactivating the Huntington gene (which produces the huntingtin protein) in a mouse results in abnormal embryo development. Supplying the mouse with either the altered (mutant) or wild-type (normal) huntingtin protein can rescue the non-huntingtin phenotype, meaning that the mouse can be brought back to a normal state. This phenomenon occurs because altered huntingtin acts like wild-type huntingtin, but gains some kind of new functions, such as a kind of “stickiness” that might make it more likely to interact inappropriately with another protein.
While touring the MacDonald lab, Altman and Fu spoke with some other postdoctoral research fellows about their research projects. Each trainee approaches HD from a different angle. For example, research fellow Elisa Fossale and intern Sony Mysore work together to prepare tissue cultures (a technique used to grow body tissue outside the body on a culture medium, a liquid or gel-like substance containing nutrients). The tissues kept in the culture medium are from the striatum of the brains of mice that carry the Huntington gene homolog (the mouse version of the gene). This brain region is known to deteriorate during the course of HD. The researchers are currently looking at differences in energy metabolism in mutant and wild-type striatal nerve cell clones. Preparing the cells for experimentation is a laborious process, generally taking about four to six hours. After this preparation, they can operationalize the changes in energy metabolism as changes in levels of the cell’s major energy carrier, ATP. These researchers have observed that ATP levels decrease in mutant cells, suggesting that metabolism is sluggish. Therefore, the cells are weakened through decreased efficiency in producing energy for their own survival. This information may be important to other researchers who use these types of cells for screening drugs that have the potential to combat decreases in energy metabolism.

Intern Sony Mysore learns how to perform a tissue culture.

HOPES team member Shawn Fu looks at striatal nerve cells under a microscope.
Other researchers and technicians work on a type of tissue culture called cell lines. Cell lines represent generations of a primary or original culture, such as a bacterial colony that arose from a single cell. Cell lines are “immortalized” biochemically so that they continue to reproduce themselves and can subsequently be used to develop tests called screening assays for potential drugs that are available from biotechnology companies and academic laboratories under contract. Once researchers have figured out the appropriate target for a drug (such as mitochondria that make ATP or nerve cell transportation machinery slowed by the effects of mutated huntingtin), they can test these chemical compounds to see which one of them really goes after the target.

Cell lines in cold storage in the MacDonald lab.
Alex Lloret, another research fellow in the lab, studies sections of the brains of transgenic mice with different genetic backgrounds to observe the formation of huntingtin in the nucleus and later formation of neuronal inclusions (NI) in the nuclei of nerve cells. Neuronal inclusions are clumps of mutated huntingtin protein fragments that result from having HD. There is generally one inclusion per nucleus in mutant mice (those with excess CAG repeats).
Lloret also looks for proteins that interact with the normal huntingtin protein. He has found that huntingtin’s function is similar to that of a scaffold, or facilitator, because it organizes groups of proteins that play various roles in signal transduction in nerve cells. He is currently in the process of mapping one end fragment of the huntingtin protein in order to understand how other protein complexes bind to it. He is also looking at HEAT repeat sequences in huntingtin and trying to determine how these sequences interact with many protein complexes.

Researcher Alex Lloret prepares sections of transgenic mouse brains for experimentation.

Sections of transgenic mouse brains are mounted on slides for viewing under a microscope.
Other methods of investigating HD include the genomic, RNA interference (RNAi), and biochemical approaches. The genomic approach entails screening various genes in a human or mouse’s body for genetic markers, measuring the transcription level (amount of transcription) of each gene, and measuring the amount of RNA produced. The RNAi approach consists, in part, of describing the normal function of the huntingtin protein. Working with HD cell models, researcher Songshan Jiang uses short interference RNA (siRNA) to stop the translation of the huntingtin protein. Nerve cells treated with siRNA do not contain huntingtin at all. Jiang then examines the phenotype of the cells, looking at the way in which they are affected by the lack of huntingtin. This kind of observation can tell him what huntingtin’s regular function is within the nerve cells.
The biochemical approach involves understanding and describing how the structural and physical properties of biological molecules (such as proteins) influence the functions of those molecules. To study these molecules, they must first be purified. One of the biggest breakthroughs in biochemistry was the introduction of chromatography, which made it possible to separate and isolate different kinds of molecules quickly and efficiently. In the MacDonald lab, a technique called gravitation chromatography is used to compare the mass of the mutant and wild-type huntingtin protein. Researcher Ihn-Sik Seong has found that mutant huntingtin is in large complexes that are even bigger (have more mass) than the complexes with normal huntingtin. They also regularly use high pressure liquid chromatography (HPLC) when studying changes in the biochemical molecules that are due the presence of mutant huntingtin in cells. Generally, protein molecules are separated according to their physical properties such as their size, shape, and affinity for other molecules. HPLC facilitates the separation of molecules under high pressure in a stainless steel column filled with a special chemical substance called a matrix. A computer controls both a pump and a means of collecting data. By using HPLC, researchers can describe various properties of mutated and wild-type huntingtin. Knowing more about the characteristics of wild-type huntingtin will give the researchers a basis of comparison for mutant huntingtin since they each have different properties.

High pressure liquid chromatography (HPLC) machines in the MacDonald lab.
Debunking the Myths
For those of us who are not familiar with the ins and outs of the scientific community, Gusella and MacDonald believe that there are many misconceptions about researcher work because then many different activities that are necessary to attack a disease like HD tend to be lumped together. In reality, these activities require quite different skills, levels of funding, and working environments.
- Myth #1: All labs are the same.Reality: There are important distinctions between academic labs (like the MacDonald and Gusella labs), industrial labs, pharmaceutical labs, clinical research labs, and clinical testing labs. All carry out different kinds of activities that are important to HD research. Generally, academic labs are responsible for discovering the biology behind a disease. Well-funded industrial and pharmaceutical labs are responsible for developing drugs that treat the disease and testing them on animals raised for that purpose. Clinical research labs are responsible for conducting clinical trials of treatments and drugs using consenting human subjects. Clinical testing labs do not do research, but instead conduct diagnostic tests for disease, in a government-approved manner, that are ordered by physicians.
- Myth #2: Researchers spend all their time researching.Reality: In fairly large academic labs like MacDonald’s, the principal researcher does little or no actual lab work (often referred to as “bench work” because the lab tables are called benches). Rather, MacDonald hires postdoctoral students and technicians to do the bench work, dividing up the different tasks and projects among them and spending time to direct their efforts. MacDonald herself spends much of her time interpreting results, strategizing for future projects, and doing administrative work, such as writing and reviewing grant proposals and sitting on scientific committees.
- Myth #3: Researchers are paid by the hospitals they work for.Reality: Researchers like MacDonald rent lab space from their institution, such as a hospital, and must do their own fundraising to obtain money for equipment and employees’ salaries. This fundraising is accomplished by applying for grants.
- Myth #4: Treatments that work on mice will usually work on humans.Reality: Most experimental drugs and treatments tested on mouse models of various diseases do not work on humans. Mice are often used to test the toxicity of the chemical compounds involved in various treatments before drugs are developed and approved for human consumption. The process of testing treatments on mice, developing drugs, getting government approval for the drugs, and putting them on the market is quite slow, has many steps, and involves making enough of a particular compound for large groups of people.Some scientists think that a larger animal model may be a promising alternative to a mouse model of HD in terms of shortening the clinical trial timeframe. A large animal model may be physiologically more similar to a human, which will give scientists a better sense of what kinds of treatments may work on humans. MacDonald mentioned that a researcher named Russell Snell at the University of Auckland in New Zealand is currently developing a sheep model of HD. Because researchers will see much more nerve cell death in sheep than in mice or rats, they might be able to get a more accurate picture of the progression of the disease in people. It will thus be easier to study the pathology of the disease in its final stages. Not only is the physiology of a sheep is closer to that of a human than a rodent’s, but a sheep model can provide other benefits in terms of HD experimentation. For example, living experiments (experiments conducted in the living animal), transplantation of cells and tissues, and protection of the nerve cells against deterioration (a process called neuroprotection) will be easier to perform.
- Myth #5: The nerve cell is healthy, and then it changes and gets sick after the onset of HD.Reality: The nerve cell in a presymptomatic person with HD is never normal. If a genetic mutation is present, then the cell is abnormal from the start. It is a common misconception that if a cell is not presently sick, it is normal. Yet, according to MacDonald, the cell merely gives the appearance of being normal by trying its best to remain healthy for as long as possible.
- Myth #6: There is one big, important change that occurs in the body to make a person sick with HD.Reality: There is certainly more than one big, important change; in fact, there is a whole series of changes! Visualizing HD in terms of a timeline allows us to think about the changes sequentially (i.e. what happens, when, and in what order during the disease cascade). MacDonald says that one of the earliest changes involves the RRS1 gene. RRS, which stands for regulator of ribosome synthesis, directs the production of this small organelle that, in turn, directs the synthesis of nuclear proteins (proteins found in the nucleus of the cell). At the beginning of the disease cascade, more RRS1 is made, perhaps because the cell needs to change its protein synthesis. Other subsequent changes take place, leading to “derangements;” for example, in energy metabolism, which lead to other changes, and so on.
- Myth #7: Scientists will find a “magic bullet” to cure HD.Reality: MacDonald thinks that it is highly unlikely that one chemical compound will work well in all people with HD. Yet she believes that HD researchers may, as a first step, be able to develop a drug to delay the onset of disease symptoms for about five years, which is the size of the effect in some individuals with a certain version of the GRIK2 (Glu6) gene. The GRIK2 gene is a genetic modifier in HD, meaning that its expression in different people can have an effect on age of onset. For instance, by manipulating this gene in a certain way, scientists can perhaps stall the onset of HD and thus shift the disease cascade in such a way that when onset actually does occur, the symptoms will be less severe.
Toward a Treatment: The future of HD research
What big questions remain in the field of HD research? Gusella said that the question, “How does mutant huntingtin trigger the disease cascade?” is, by far, the most important one because learning about the root cause of the disease will help researchers intervene early in the cascade before the onset of symptoms. Secondary questions include:
- What is the normal function of the huntingtin protein?
- Is there a point at which the disease pathway becomes independent of its trigger?
- What are the phenotypes in tissues other than the brain of a person with HD?
- Why are cancer rates lower in people with HD?
- What is the overall effect of HD on metabolism (not just in certain fat cells)?
- What is the origin of the chromosome that gives rise to new mutations?
- What produces the long length of the HD allele?
- Why don’t nematodes (a type of worm) have a Huntington gene? (Nematodes are multicellular – made up of many cells – just like humans and mice.)
MacDonald discussed what she believes will be an important future discovery: the development of a biomarker to measure the progression of HD in a patient. She thinks that a rating scale that measures the functional decline (the physical deterioration) of people with the disease may prove to be important. She would also like to see a quantitative test that reflects the state of a person’s disease – not just for HD, but for Parkinson’s and cancer, as well. Unlike MacDonald, Gusella thinks that a biomarker would not be particularly meaningful; rather, he would like to see researchers direct their efforts toward developing a marker that could indicate a reversible disease state.
What level of specificity within the body will be targeted by a breakthrough treatment – the cell, gene, protein, DNA, or RNA? After a drug, which would be their first choice, MacDonald and Gusella said that RNA, but not DNA, is a promising level for a treatment. RNA is promising in terms of delivery to nerve cells (siRNA is delivered to the cells via the RNAi technique). At the protein level, it is possible to deliver normal huntingtin to the nerve cells in a similar way. However, the only drawback is that the technology involved in these processes is relatively unknown and underdeveloped. On the bright side, scientists and doctors can target the mutant huntingtin with small-molecule pharmacological drugs that are already available. At the level of the nerve cell, it seems promising to target treatments to nerve cell-nerve cell interactions at a distance from the trigger of the disease cascade. Unfortunately, this may be difficult due to the fact that the trigger is still unknown. On the other hand, targeting drugs to genetic modifiers would be a faster route than targeting nerve cell-nerve cell interaction – if such a route were available.
Conclusion
Gusella, MacDonald, and their fellow researchers in the United States and around the world will continue to work diligently to find a treatment for HD. Much has already been accomplished by dedicated research teams, and many more breakthroughs are soon to come with advances in technology and the development of more accurate animal models of the disease. Once researchers identify the trigger, they can develop an effective treatment that stops the disease cascade early enough to prevent the onset of symptoms.
In the meantime, how can people with HD help the researchers in their quest for a breakthrough? MacDonald said that they can ask their doctors if they can participate in ongoing research, such as clinical trials, genetic studies, or observational studies. To learn more about current research, people with HD should read informative websites like the Huntington Study Group site. MacDonald added that newly diagnosed people should ask their doctors for something to read (a booklet or pamphlet) about managing the symptoms of HD, such as A Physician’s Guide to the Management of Huntington’s Disease, Second Edition by Drs. Adam Rosenblatt, Neal G. Ranen, Martha A. Nance, and Jane S. Paulsen.
“Research is a winding journey from the starting point to where you want to end up,” MacDonald said of the experience of working on HD (or any scientific phenomenon). It has been a long road, and she and Gusella still have miles to go, but they can see their destination – a much-anticipated treatment – on the horizon.

L-R: HOPES team member Shawn Fu, Dr. Marcy MacDonald, and HOPES team member Taylor Altman
-T. Altman, S. Fu, M. Stenerson, 12/07/04
More
HDSA Center of Excellence at Columbia University
New York State Psychiatric Institute
New York, NY
In the summer of 2005, HOPES researchers Jia Hou and Clare Tobin visited the HDSA (Huntington’s Disease Society of America) Center of Excellence at Columbia University in New York, NY. Hou and Tobin would like to thank everyone at the center for taking the time to speak with them on an especially busy day. They would also like to thank the patients who gave such candid interviews and permitted us to observe their evaluations. To visit the Columbia center’s website, click here.
Introduction
The HDSA Center of Excellence at Columbia is a highly specialized program. It is primarily a clinical facility, but medical research is a very important part of the center’s mission as well. When patients come to the center, they are screened as possible participants for the many HD-related research studies that are conducted elsewhere in the hospital. Patients may choose whether or not to participate; either way, they receive high quality care.
Because HD is such a complex disease to manage, the center provides many different kinds of specialists and medical services. Hou and Tobin were able to meet several members of the center’s interdisciplinary team, including:
Karen Marder, MD, MPH – Neurologist (and Center Director)
Mark Groves, MD – Psychiatrist
Jeanne Thomson, MS, CCC-SLP – Speech Therapist
Ashwini Rao, Ed.D, OTR – Occupational (and Physical) Therapist
Deborah Thorne, LCSW – Social Worker
Jennifer Williamson-Catania, MS – Genetic Counselor
Paula Leber, MA – Research Assistant
To read more about these staff members and the rest of the staff, click here.
A significant part of the center activities revolve around people who are at risk for HD, but are not symptomatic. These people have a family history of HD, but do not know if they carry the HD allele. One way to determine this is with predictive genetic testing. (For more information on testing, click here). For issues surrounding predictive testing, Hou and Tobin spoke with Jennifer Williamson-Catania, the center’s genetic counselor. More information on this topic will be discussed later in the chapter. In the meantime, the focus will be on the organization of the center and how it helps those with HD.
The Clinic
HOPES visited the Columbia center on August 19th, 2005 to observe one of their monthly clinic days. On these special clinic days, many patients come into the center. The team of specialists that comprise the center’s comprehensive care program will then re-evaluate each patient’s disease progression. On their first visit, patients meet with the entire team of specialists, and then usually come in two times each year after that. Patients may come in more often, depending on the progression of their condition. The medical team consists primarily of a psychiatrist, neurologist, speech therapist, occupational/physical therapist, and social worker. The team meets with each patient (and usually his or her primary caregiver) to examine the patient’s current health status. The patient may then meet with individual specialists to discuss specific adjustments to treatment and lifestyle. For example, speech therapist Jeanne Thomson may recommend getting speech therapy a few times a week for a number of months. Speech therapy can help with communication, especially over the phone, as well as problems with eating. It is very important to address eating issues because coughing, choking, and aspiration pneumonia are serious concerns for people with HD. (For information on aspiration pneumonia and other complications of HD, click here). For patients that do not live near the center, Ms. Thomson refers them to therapists who work in their area. There is a similar procedure for physical therapy and other forms of therapy. The team may prescribe medications that alleviate symptoms, but there is no medication that can treat the disease itself. In general, the team recommends against taking any kind of dietary supplements for the purpose of treating HD.
For all patients, care at the center is free of charge. This situation certainly eases the burden on families, makes it possible for patients to receive the care that they need without financial constraints, and better protects confidentiality by avoiding involvement with insurance companies. However, patients still face other costs, such as hiring home health care aides or receiving treatment for other conditions. For these reasons, the center is quite concerned with health insurance issues. The people at the center help patients figure out how to afford the other aspects of their care by considering alternative resources, such as Medicaid, Medicare, and disability insurance.
Resources and Support
The center provides many resources beyond those of standard clinical care. There are many specialists available for consultation, as well as other HD patients for mutual support. The center has a large body of HD-related literature for patients to read. There is an at-risk support group and an early-to-middle stage patient support group that each meets once a month at Columbia. Other support groups meet at different nearby New York locations. The center takes part in many events throughout the year that raise money for HD research, from amaryllis bulb sales to a BMW sweepstakes and “Hoop-a-thons.” Hou and Tobin were fortunate during their visit to meet a young boy and his family who helped raise a large sum of money for HD research; they were inspired to coordinate such an effort because of a neighbor that had been affected by HD. (For more information on upcoming events at the center, click here).
Another way that the center improves the lives of people with HD is through their annual camp, which was started in 1993. During the summer, 14-18 patients spend a week in Cornwall-on-Hudson, NY with camp staff. Many members of the camp staff also work at the Columbia center. At camp, everyone takes part in all sorts of activities, from singing to horseback riding to pool volleyball. This special week gives patients a chance to have fun, spend time with others who understand what it is like to have HD, and bond with people from the center. The week also provides a well-deserved vacation for caregivers. (For more information on the HD Summer Camp Program, click here).
Research
Though the Columbia center is primarily a clinical facility, they also conduct important medical research. Patients often choose to become involved in research studies or clinical drug trials to help advance scientific knowledge about HD. Because HD tends to run in families, many patients are particularly concerned about finding effective treatment, even if not for themselves. During the HOPES visit, Hou and Tobin were able to observe the preliminary stages of a research project that uses fMRI scans to try to find early changes in the brain that are associated with HD. The study compares the scans of presymptomatic and symptomatic individuals (people who have HD) and controls (people who do not have HD). Several patients from the center volunteered for the scans, and several of their spouses volunteered as controls.
Hou and Tobin were also invited to sit in on a lunchtime presentation given by a Columbia medical student who had been researching HD. First, he discussed dietary supplements and HD. There have been many reports about the effectiveness of various supplements in combating the progression of HD. Sometimes these reports are exaggerated by people outside of the medical community who are well-meaning but impatient for a cure. The student’s presentation showed that, while some supplements show promise, there is no evidence of benefits at this time. This is why the center’s team recommends against taking supplements for the treatment of HD. (For more information on dietary supplements, click here). Hou and Tobin were proud to note that the information in the presentation had already been evaluated and posted on the HOPES website prior to their visit.
The second part of the presentation was about driving and HD, a common and serious concern for patients and their families. At a certain point in the progression of the disease, it may no longer be safe for people with HD to continue to drive. However, this can mean a loss of independence and self-sufficiency, which makes it a very difficult decision. If there is concern about a patient’s ability to drive, the team may recommend an independent driving evaluation. Though many people with HD continue to drive successfully for a long time, driving and HD is a very important issue which needs to be more widely discussed in the HD community.
Bridging the Medical and Social Aspects of HD Care
In addition to managing the physical symptoms of HD, a good clinical facility also needs to consider the psychosocial and overall well-being of its patients. What sets the Columbia center, and other Centers of Excellence, apart from standard treatment is how the teams there provide comprehensive care. In addition to making a diagnosis, assessing symptoms, and recommending treatments, the clinical team considers factors such as a patient’s access to continuing care, sources of funding, and concurrent medical conditions.
The center’s team also helps patients make informed decisions about various aspects of their lives, while respecting their autonomy. For example, it can be very difficult for someone with HD to give up daily activities like driving, but these decisions eventually need to be made. Yet it is difficult to determine exactly when to make these decisions, given the gradual progression of the disease. Comprehensive, individualized care from the team can help people with HD and their families make important decisions.
Families affected by HD must also make decisions about predictive genetic testing. Genetic testing reveals whether a person has the HD allele, and thus whether he or she will eventually get HD. In order to make an informed decision, people need to have as much knowledge and guidance as possible. The Columbia center’s genetic counselor, Jennifer Williamson-Catania, informs at-risk individuals about the facts of genetic testing and the meaning of HD risk factors. She also makes them aware of reproductive options, such as pre-implantation genetic diagnosis (PGD). (For more information on PGD, click here). Though Ms. Williamson-Catania lays out all of the potential outcomes and alternatives to be considered, she emphasizes that she does not provide any easy “answers” or “solutions.” She is careful not to let her personal values influence the advice she gives, as the decision to be tested is a very serious one, both for the individual and for his or her family. The possibility of a genetic test only further shows how important a full range of support is for families with HD. (For more information on testing, click here).
In Conclusion
Over the course of their visit, Hou and Tobin were impressed with the devotion and versatility of the center’s team. It was a busy day and the center was sometimes caring for four different patients at once. Yet the team still took the time to patiently answer questions from the HOPES researchers. While Ms. Williamson-Catania showed Hou and Tobin the fMRI room and discussed the difficulties of genetic counseling, she also managed to hurry back to the reception room intermittently to greet patients that had just arrived. Watching the team members in action showed the energy, attention, and empathy they had for their patients. Indeed, the patients had only positive things to say about the care and the sense of community that they experience at the center. It was clear to the HOPES researchers that the Columbia center is a model for the comprehensive care of HD.
More
The following chapter aims to compare Huntington’s disease to other neurological diseases such as Alzheimer’s disease and Parkinson’s disease.
Risk Factors
HD is caused by a mutation in the Huntington gene, which lies on chromosome 4. A certain sequence of DNA (C-A-G) of the Huntington gene is repeated multiple times. Generally, if a person has 35 or fewer copies of CAG on a particular segment of the Huntington gene, the person will not get HD. However, if he or she has 40 or more copies, he or she will get the disease. The greater the number of repeats, the more likely it is that the person will develop symptoms and the greater the chance that these symptoms will occur at a younger age. Every child of a parent with HD has a 50% chance of inheriting the disease, and the disease may occur earlier and more severely in each succeeding affected generation because the number of repeats canincrease. For more info on inheritance, click here.
gene altered neurons base ganglia control nucleus
Like Huntington’s disease, family genetics present a significant risk factor for inheriting Alzheimer’s disease. A study performed by Erasmus University Medical School in The Netherlands found that the risk of developing Alzheimer’s disease for those with at least one affected immediate relative was 3.5 times greater than those with no affected relatives. The study also showed a significant association between Alzheimer’s disease and family history of Down’s Syndrome, as well as an increased risk in with a family history of Parkinson’s disease. Perhaps the most significant risk factor of Alzheimer’s disease is age. Although there is variability in the age ofHD symptom onset, the incidence of Alzheimer’s increases with age, doubling every five years from 1% at 60 years to as many as 50% for those over 85 years of age. This is because normal aging is associated with altered protein metabolism, a process that often leads to the degradation of brain cells and the formation of abnormal clumps of protein in the brain over time. Other risk factors for Alzheimer’s disease include Down’s syndrome, untreated chronic hypertension, high cholesterol, and sustained head injuries.
Age is also the main risk factor for developing Parkinson’s disease. A subtype of Parkinson’s disease called young-onset Parkinson’s disease affects those younger than 40; however, most of those affected do not experience symptoms until after age 55. Although controversial, researchers now think genetics may play a rolein the development of the disease. The recent discovery of an abnormal protein called a-synuclein in an Italian family with many Parkinson’ssufferers has contributed to the understanding of Parkinson’s disease. As of early 2002, there were nine genetic abnormalities that had beenassociated with Parkinson’s disease. New studies show that having one close relative with Parkinson’s may increase the chances of developing Parkinson’s three or four-fold, and having two or more relatives may increase the chances ten-fold. However, most often a definite family history is not present for most patients. In these sporadic cases, agenetic predisposition may still play a role by increasing the chance of getting the disease when patients are exposed to possible environmental risk factors such as certain pesticides and herbicides. Other risk factors that may contribute to Parkinson’s disease include reduced estrogen levels and low folate levels (see section onnutrition).
Neurobiology: Neuromotor Comparisons
As addressed in the HD Neurobiology chapter, the part of the brain most affected by HD is a group of nerve cells (neurons) at the base of the brain known collectively as the basal ganglia. The basal ganglia is responsible for the muscle-driven, motor movements of the body. As the cells in this area die, a person with HD experiences uncontrollable muscular movements likened to fidgetiness or nervous restlessness.
Similarly, Parkinson’s disease patients experience uncontrollable movements due to the disease’s effects on a specific area of the basal ganglia called the substantia nigra. The substantia nigra produces a chemical called dopamine, which is a neurotransmitter. Neurotransmitters are special chemicals that help neurons communicatewith each other. Dopamine and another neurotransmitter, acetycholine help to control our movements. In Parkinson’s disease, the neurons inthe substantia nigra gradually die off, which causes less dopamine to be made. With less dopamine than normal, there is an imbalance between dopamine and acetylcholine (see figure). This imbalance causes the nerve cells to fire out of control, leaving patients unable to direct their movement in a normal manner.
In Alzheimer’s disease, neurons in the brain and the spaces between them become clogged with protein deposits called beta amyloid plaques and neurofibrillary tangles. Even in people who don’t have AD, plaques and tanglesdevelop as part of the normal aging process. However, in people with Alzheimer’s disease, there are many more plaques and tangles. Plaques are dense, mostly insoluble (cannot be dissolved) deposits of protein and cellular material outside and around the neurons. Tangles are insoluble twisted fibers that build up inside the nerve cell. When neurons are clogged with tangles, and the spaces between neurons are clogged with plaques, the transmission of nerve impulses from one neuron to the next does not happen properly. As a result, the brain cannot perform mental functions such as remembering and thinking. There are other senile dementias (like Multi-infarct Dementia) that present like Alzheimer’s disease but have very different causes and are not comparable to HD. Though they many share certain neurobiological properties, these are distinctive conditions.
Though Huntington’s, Parkinson’s, and Alzheimer’s disease are caused by unique cellular processes, all three diseases are facilitated by the inability of neurons to communicate with each other. Gradual neuron death in people with HD hinders the ability of neurons to communicate. For people with PD, neurons die, causing an imbalance of dopamine and acetylcholine and the uncontrolled firing of nerve cells. Finally, in people with AD, plaques and tangles inhibit neurons’ ability to communicate.
Neurobiology: Emotional/Cognitive Comparisons
Disclaimer: Despite many similarities, these cognitive and emotional signs present at different stages of the disease in different people. A person with HD may very well maintain healthy cognitive functioning throughout the remainder of his/her life.
The symptoms of Huntington’s disease are both behavioral and cognitive. Symptoms are the direct result of neurological changes in the brain. Apathy is one of the most common behavioral symptoms of HD due the death of nerve cells controlling “emotions” in the brain. Deterioration of a certain area of the brain called the caudate nucleus causes HD sufferers to be unable to control intensities of emotion, and makes them more likely to experience frustration, irritability, and aggression. For more on behavioral symptoms associated with HD go here.
In addition to behavior symptoms associated with HD, many cognitive changes also arise with the onset of Huntington’s disease due to neuronal damage. A patient’s ability to initiate a conversation and to communicate is altered due to degeneration in the brain. Furthermore, an individual suffering from the cognitive symptoms of HD may have memory, problem solving, and judgment difficulties. Tasks that were once simple are difficult for an HD patient to perform efficiently. An HD patient also experiences difficulty with visual spatial impairment, awareness, and organization. For more on the cognitive symptoms associated with HD, go here.
Similarly, patients of Alzheimer’s disease may experience both behavioral and cognitive changes at different stages of their disease process, many which are similar to HD. Difficulty with the acquisition of new information is generally the most salient symptom to emerge in patients with AD. Whereas learning new information for HD patients is disorganized and slow, Alzheimer’s patients experience rapid forgetfulness and an inability to store information. Several studies have demonstrated that people with AD lose more information over a brief delay than other patients with disorders that involve amnesia or dementia. Though at first their symptoms may be mild, people in the later stages of AD may forget how to perform simple tasks, like brushing their teeth or combing their hair. They neglect to bathe, or wear the same clothes over and over again while insisting that they have taken a bath or that their clothes are still clean. They can become lost on their own street, forget where they are and how they got there, and not know how to get back home. Eventually, patients need total care because they are unable to think clearly and perform tasks for daily living.
Another similarity to HD is that Alzheimer’s patients lose their initiative to perform normal activities or to engage in activities they used to enjoy. They often become very passive, sitting in front of the television for hours and sleeping more than usual. Furthermore, Alzheimer’s patients can experience rapid mood swings for no apparent reason, and their personality can vary from becoming extremely confused and suspicious to being fearful or dependent on a family member. They also may see, hear, smell, or taste things that are not there. Finally, like those with HD, Alzheimer’s patients sometimes exhibit poor judgment, which creates safety issues when left alone. They may wander and risk exposure, accidental poisoning, falls, self-neglect, or exploitation.
For patients with Parkinson’s disease, the most prominent symptom is tremor. Tremor often starts in one extremity and worsens with precipitating factors such as stress, fatigue, and cold weather. The tremor associated with PD occurs predominantly at rest, and results in the slowness of a patient’s movement (also known as Bradykinesia) A delay in initiating movements develops due to the brain’s inability to transmit necessary instructions to the body at a normal rate. Parkinson’s patients often report difficulties in performing activities of daily life, such as dressing, walking, and doing household chores. Symptoms that appear later in the progression of the disease include poor balance and the inability to swallow. Upon walking, a Parkinson sufferer has a decreased or non-existent arm swing, short shuffling, and difficulty negotiating turns. Another major symptom is rigidity, characterized by increased tone and stiffness in the muscles; rigidity is responsible for a Parkinson patient’s sometimes mask-like facial expressions and stooped posture.
As with HD and AD, depression is commonly seen in the early stages of Parkinson’s disease. It is estimated that about half of people with Parkinson’s may suffer from depression. This is thought to be not only a reaction to the diagnosis, but rather an intrinsic part of the disease process. Also, as with HD, Parkinson’s disease causes anxiety and can cause panic attacks. Symptoms of anxiety include breathlessness, sweating, chest discomfort, choking, and dizziness. In severe cases, patients may have feelings such as the fear of dying or the fear of going insane. Also, about 15-25% of individuals with Parkinson’s disease will suffer from memory and cognitive deficits similar to those of Huntington’s disease patients. Mild cognitive deficits are common in Parkinson’s and are characterized by a lack of flexibility in thought, difficulty in learning new information, and impaired visual-spatial skills. Short-term memory deficits are common and may progress to more severe memory deficits. Language skills are relatively spared although some studies have found a mild impairment in naming. Higher executive function (abstract thinking, planning abilities, judgment, and initiative) is often affected in patients with Parkinson’s disease as well.
Treatments
Currently there exists no cure for any three of the neurological diseases. As with Huntington’s Disease, treatments for Alzheimer and Parkinson’s can be split into two distinct categories: treatments that target the specific mechanism of the disease, and palliative treatments (eg those that lessen symptoms but do not cure). For HD, mechanisms which are targeted include protein aggregation, inflammation, and free radical damage (See treatments section).
As of this writing (Jan 04), there are five FDA-approved drugs that can control symptoms and slow the progression of Alzheimer’s disease. Four of these drugs, Cognex, Aricept, Exelon, and Reminyl belong to a class of drugs known as cholinesterase inhibitors. Each drug acts in a different way to slow the metabolic breakdown of acetylcholine, an important brain chemical involved in nerve cell communication, and to make more available for communication between cells. Those suffering from AD have low levels of acetylcholine, and the medication helps to slow the progression of cognitive impairment and is most effective for patients in the early to middle stages of AD. The fifth drug, Namenda (memantine), is the first drug approved for the treatment of moderate to severe AD. Namenda shields brain cells from overexposure to another brain chemical called glutamate, excess levels of which contribute to the death of brain cells in people with Alzheimer’s. Although all five drugs have all been shown to modestly slow the progression of cognitive symptoms and reduce problematic behaviors in some people, at least half of the people who take these drugs do not respond to them. While the overall treatment effect of these medications is modest, studies show that, when they do work, they can make a significant difference in a person’s quality in life and day-to-day functioning.
Research is now focused upon prevention trials which try to stop the disease process from happening in the first place, and a number of studies are underway to test the effectiveness of various therapies in people without symptoms or who have only slight memory problems. Some of these studies are examining estrogen and various classes of anti-inflammatory and antioxidant chemicals. Research has shown that vitamin E and other antioxidants may slow the progression of AD in some people, although the overall impact is minimal. Research also suggests that ginkgo biloba, an extract made from the leaves of the ginkgo tree, may be of some help in treating AD symptoms. However, there is no evidence that ginkgo will cure or prevent AD. (For more on ginkgo biloba, click here.)
Palliative medications that can control depression, anxiety, agitated behavior (including aggression, hyperactivity and combativeness) and psychotic symptoms can help patients in the middle stages of AD. The medications prescribed for these symptoms are not specifically designated for AD, but they may be considered as part of the treatment plan by the supervising physician. Generally, medications for these symptoms are considered when non-medicated alternatives have failed and/or these symptoms put the AD patients, or others, in danger.
The purpose of all medicines for Parkinson’s disease is to help control tremor, movement, and balance to maintain daily activities. One of the mechanisms targeted by Parkinson’s medications includes the interactions of dopamine, a neurotransmitter (chemical messenger) that affects brain processes by allowing nerve cells to communicate with one another in the brain. Scientists have determined that people with late PD have lost more than 80 percent of dopamine-producing cells in the substantia nigra, an area deep within the brain. Normally, these cells communicate with other brain cells in the nearby striatum via dopamine. Thus, without dopamine, the striatum can’t send out certain messages and the symptoms of Parkinson’s ensue. Levodopa, also called L-dopa, was the earliest treatment discovered for Parkinson’s disease. L-dopa is a method of dopamine replacement therapy; it is turned into dopamine in the brain to supplement the cells that are producing less.
Another group of medications fit into the category of dopamine antagonists, drugs that bind but don’t stimulate dopamine receptors. Antagonists can prevent or reverse the actions of dopamine by keeping dopamine from attaching to receptors; they help improve control of various body movements, which begin to slow or become irregular in early Parkinson’s disease. Dopamine antagonists work by copying the effect of the neurotransmitter dopamine, proving effective in people with Parkinson’s disease who are losing their dopamine-producing cells. By doing this, dopamine antagonists can help people maintain their daily activities. Furthermore, anticholinergic drugs can be used to treat mild symptoms of Parkinson’s disease. Anticholinergic drugs block a neurotransmitter that affects dopamine so that more dopamine is available in the brain. Other pharmacological medications exist to treat Parkinson’s, and they too usually involve the mimicking or replacement of dopamine.
Prognosis
As discussed elsewhere in this website, HD is a progressively debilitating disease with no known cure. The person with Huntington’s disease may be able to maintain a job for several years after diagnosis, despite the increase in disability. Loss of cognitive functions and increase in motor and behavioral symptoms eventually prevent the person with HD from continuing employment. Ultimately, severe motor symptoms prevent mobility. HD is usually fatal within15 to 20 years. Progressive weakness of respiratory and swallowing muscles leads to increased respiratory infection and choking, the most common causes of death. (For more information about the complications of HD, click here.) However, not all patients with Huntington’s disease progress at the same pace and are equally affected. The number of repeats may determine severity. There are people with a low number of repeats that have mild abnormal movements later in life and progression is slow whereas others with a large repeat length who are severely affected at a young age.
Although different in many ways, Alzheimer’s disease is also an incurable and progressively debilitating disease that can vary widely in its progression. Some people have a very precipitous course and go downhill rapidly, while others remain stable for a long time. For some, the disease only for the last 5 years of life; others may have it for as many as 20 years. A study of the prognosis of AD at the University of Massachusetts Medical School suggests that initial degree of severity (“how far”) rather than the variation in the rate of progression (“how fast”) best predicts prognosis in the early to intermediate stages of Alzheimer’s disease. Total disability is common in people with Alzheimers, and the most common cause of death is infection or a failure of other body systems.
Predicting disease progression for Parkinson’s disease is difficult because of the wide spectrum of disease types. Again, the course and prognosis of this disease vary according to the individual. Without treatment, PD causes severe disability or death in 25% of patients within 5 years, 65% of survivors after 10 years, and 89% of survivors after 15 years. However, with treatment, the life expectancy of people with PD without an accompanying dementia is nearly normal. The mean time from diagnosis to death in treated PD is 14 years. Death is usually due to complications of immobility, such as pulmonary embolism (blood clot in the lungs) or aspiration pneumonia (lung infection from regurgitated stomach contents).
More
Huntington’s Disease (HD) is not fatal in itself. People with HD have a shorter life expectancy and die of other life-threatening complications related to this disease. Pneumonia and heart disease are the two leading causes of death for people with HD. Additionally, HD patients have higher incidence of choking and respiratory complications, gastrointestinal diseases (such as cancer of the pancreas), and suicide than the non-HD population. Why are HD patients more prone to the above complications than the rest of the population? This chapter aims to answer that question and draw connections between the symptoms of HD and the most common causes of death (see the table below). Although researchers have not explicitly proven these links in every case, the following information hopes to demonstrate a logical connection.
| Primary cause of death (in rank order) |
Persons (total=182) |
Percentage |
Pneumonia
Other respiratory diseases |
93
5 |
51.1
2.7 |
Myocardial infarction/degeneration (heart attack
Congestive cardiac failure (heart failure)
coronary disease
Other diseases of the cardiovascular system |
5
18
11
2 |
2.7}
9.9}
6.0}19.7
1.1} |
| Unspecified Huntington’s-related causes |
23 |
12.6 |
| Vascular lesions of central nervous system |
10 |
5.5 |
| Non-vascular lesions of central nervous system (e.g. meningitis) |
4 |
2.2 |
| Genito-urinary diseases (e.g. kidney failure) |
5 |
2.7 |
| Gastro-intestinal diseases (cancer of the pancreas) |
3 |
1.6 |
| Suicide |
3 |
1.6 |
| Reed TE, Chandler JH, Hughes EM, et al. Huntington’s chorea in Michigan: I. Demography and Genetics. Am J Hum Gen 1958; 10: 201-225. |
One of the chief symptoms of HD is the inability to produce coordinated movements. In the latter stages of the disease, this problem becomes more pronounced to the point that people have difficulty swallowing. Although it is so common that we hardly think about it, swallowing is actually a complex series of movements by muscles in our throat to ensure passage of food into the esophagus (gastrointestinal tract) rather than the trachea (respiratory tract). As a result of these movements, the epiglottis, a flap that acts as a valve in our throat, prevents food from entering the airway. People with HD often lack this coordination, and food will accidentally enter the respiratory tract, leading to choking. Moreover, when food particles manage to get into the trachea (the “wind pipe” leading to the lungs), instead of the esophagus (the “food pipe” leading to the stomach), the lungs can become infected and cause what is known as aspiration pneumonia.

Although pneumonia is relatively common among people in the general population, it is only fatal in about 5% of these cases. However, pneumonia is much more dangerous in people with compromised immune systems. Researchers have demonstrated that stresses imposed on a person for prolonged periods of time can severely damage the body’s ability to ward off diseases. The physical, cognitive, and psychiatric symptoms of HD add a great deal of stress to everyday life for these patients (for more information on these symptoms, click here). As a result, their immune systems are compromised and diseases such as pneumonia are therefore more likely to result in death. For instance, in a long-term study conducted from 1952 until 1979 in Victoria, Australia, researchers found that more than 51% of patients with HD died from pneumonia.
The increased physical and emotional stress associated with HD can cause other problems as well. Chronic stress has been linked to high blood pressure, increased risks for heart attacks, and tumor growth. In addition, although studies have shown that suicide is not a leading cause of death for HD patients, suicide rates are higher than among the rest of the population. This is probably due to a combination of factors, including neuropsychiatric changes induced by HD and the added stress of daily life.
Although researchers have yet to find a cure for the disease, people with HD can take measures to prolong their lives. For example, extra care should be taken when eating to prevent choking and pneumonia caused by food going the wrong way. Regular exercise and sleeping in an elevated position can reduce the risk of respiratory infections. Patients can also maintain a healthy diet and reduce or eliminate other risk factors for heart disease, such as smoking and alcohol, from their lives.
More
In July 2008, HOPES researcher Tiffany Wang visited the Huntington’s Disease Society of America Center of Excellence located at the Hennepin County Medical Center (HCMC) in Minneapolis, Minnesota. The Center of Excellence serves approximately four hundred Huntington’s Disease (HD) patients and families from Minnesota, North Dakota, South Dakota, Northern Iowa and Western Wisconsin. Although it was not formally designated as a Center of Excellence at its start, the HD clinic at HCMC has provided care to HD patients since it was founded in 1978. For more about HDSA Centers of Excellence please click here. Today, under the direction of Medical Director and neurologist Dr. Martha Nance, the HD clinic continues to provide patient care through an expanded program. The clinic is staffed by a multidisciplinary team of specialists, some of whom have worked with the HD clinic for over a decade. HOPES had the opportunity to speak with several of the staff members:
- Susan Braun-Johnson, P.T. Physical Therapist
- Sally Gorski, M.A. Speech-Language Pathologist
- Carol Ludowese, M.S. Genetic Counselor
- Mary Morgan LaGorio O.T.R. Occupational Therapist
- Stacey Payerl, R.D. Clinical Dietician
- Dawn Radtke, R.N. Clinical Research Coordinator
- Lena Ross, M.S.W. Social Worker
- David Tupper, Ph.D. Neuropsychologist
Patient Care
The HCMC Huntington’s Disease clinic is located in the Center’s Neurology department and provides patient care through weekly in-house admittance, monthly HD clinics and visits to long-term care facilities. Some of the clinic’s HD patients represent families that have been with the HCMC clinic for three generations. Generational patient care facilitates strong understanding of patient histories and good patient-physician relations.
Every Wednesday, neurologists Dr. Martha Nance and Dr. Scott Bundlie provide care to HD patients through in-house admittance at the medical center. These appointments with the neurologists are generally for newly diagnosed HD patients, genetic testing consultations and medication changes.
Patients who require other services are advised to visit the clinic during the monthly HD clinic days which occur one Wednesday per month. On these days, a multidisciplinary team of specialists offers services including consultations in neurology, physical therapy, speech-language pathology, genetic counseling, occupational therapy, nutrition, neuropsychology, and social services. Dr. Nance lauds the clinic days as being particularly helpful because patients and their families can see several different physicians and specialists without the hassle of scheduling several appointments or coming to the hospital for multiple visits. HOPES members were able to visit Minneapolis on one of the HD clinic days to see how they are run.
Before the HD clinic opens, the staff holds a meeting to determine the needs of incoming patients. Dr. Nance reviews relevant medical history and highlights concerns that may arise for each of the approximately ten patients who visit on clinic days. By holding the pre-clinic meeting, the staff members are prepared for more meaningful interactions with patients. Following the meeting, the specialists disperse to tend to the patients. Dr. Nance greets most patients personally, inquiring about how the patient has been since his or her last visit. Check-ups for the HD patients are usually scheduled for every three months, so most patients have news to tell Dr. Nance whether it is about serious new symptoms or simply a recent family vacation.
In many instances, the patient’s family and caregivers will accompany them to the clinic and can give insight into how the patient is coping with HD. Family members, particularly siblings and children of HD patients, may want to consider genetic testing. Genetic counselor Carol Ludowese talks with family members about their options for genetic testing. She can also give advice to younger family members about options like pre-implantation genetic diagnosis that would enable them to have children without passing on the genes for HD. For more information about genetic testing click here.
Following each of her consultations, Dr. Nance will report back to the rest of the HD clinic team who will go into the rooms to see the patients. The other specialists will then tailor visits and consultations to the needs of the patients and their families. In addition to finding out how the patients are doing holistically, Dr. Nance also asks more specific questions depending on the condition of the patient. Many of these questions are about topics that can be more thoroughly addressed by one of the specialists on the team.
Dr. Nance often asks about the patient’s weight and eating habits. One of the common symptoms of HD is weight loss, and in the later stages of the disease patients’ weights can fall significantly below healthy levels. If patients are showing drastic weight loss clinical dietician Stacey Payerl offers advice on how to maintain a healthy weight. Her recommendations often extend beyond what kinds of foods to eat to how caregivers can encourage food intake by making eating more enjoyable for the patient.
Speech-language pathologist Sally Gorski can also help patients who are having trouble maintaining a healthy weight. Patients find swallowing becomes more difficult as HD progresses, so she can administer a swallow exam to determine what kinds of foods are safe for a patient to eat without choking. To increase calorie intake and overcome swallowing difficulties, physicians often recommend that a feeding tube be inserted into the stomach. Dr. Nance emphasizes that whether or not a patient wants to have a feeding tube is an important issue to discuss early on because when the time comes to make the decision, the progression of HD may make it too difficult for patients to make the decision or convey their wishes to their families. Gorski can help patients and their families learn more information to help decide whether a feeding tube is right for them. Making the decision to have a feeding tube falls under the category of advanced directives, which Dr. Nance thinks are important to bring up to patients even during beginning stages of the disease.
While engaging the patients and families in conversation, Dr. Nance often asks patients to complete several motor tasks. Some of these actions include sticking the tongue out, walking a short distance, tapping the index and middle fingers against the thumb, and looking in different directions without moving the head. Watching the patients’ performances helps Dr. Nance evaluate which stage of HD they are in according to the Unified Huntington’s Disease Rating Scale (UHDRS). Physical therapist Susan Braun-Johnson and occupational therapist Mary Morgan LaGorio can provide more extensive advice to patients about their physical and motor symptoms, as well as ways to cope with these symptoms. For example, assistive devices can help in daily activity and changes within the home, such as installing additional bathroom equipment, can maximize safety. For more information about assistive devices and physical therapy click here.
Patients are also asked about their mental status and, if they are still formally employed, whether they are experiencing any difficulties at the workplace. These questions can help determine how behavioral and cognitive symptoms are progressing. If more thorough examination is needed, neuropsychologist Dr. David Tupper can administer several neuropsychological tests to determine how a patient’s brain is functioning. Results from these tests not only help members of the HD clinic better understand their patients, but they can also be important for determining qualifications for social security disability.
Questions about disability determination and other topics related to social services can be answered by the social worker Lena Ross on the HD clinic team. She can advise HD patients about resources within the community. One of the recurring topics that Lena receives questions about is health insurance. Given the complex nature of health insurance, patients along with families and caregivers often find it difficult to understand how HD patients can manage their healthcare costs. Lena can also answer questions regarding care for the patients who are progressing into the later stages of HD. Many family members of HD patients find it difficult to balance their busy lives with caring for a loved one with HD. It is important for families to recognize that HD patients can receive meticulous care without becoming a burden for their family at home. If a patient begins showing more severe symptoms, it may be safer to consider other options, such as a long term care facility. In this regard, Lena helps HD families learn about and weigh the options that are available to them.
At the end of the day, the HD clinic team meets again to discuss the status of the patients. Many patients are scheduled for another visit in three months or, if necessary, follow-up visits within a shorter amount of time. Although official reports are documented for each patient, the post-clinic session helps the HD clinic staff familiarize themselves with the patients for more individualized care.
For patients who are in the later stages of HD and cannot go to the clinic in Minneapolis, staff from the clinic visit local long-term care facilities to provide care to HD patients. The HCMC Center of Excellence is affiliated with the Good Samaritan Society – University Specialty Center also located in Minneapolis. The facility provides long-term care to patients with several chronic diseases and has a unit for HD patients that can care for up to fifty patients. Dr. Nance organizes monthly visits to the Good Samaritan Society patients. Whether at the HCMC or the Good Samaritan Society facilities, the HD clinic continues to provide comprehensive and personal care to its patients.
Research
Dawn Radtke, Clinical Research Coordinator, spoke with HOPES about the research that is conducted at the HCMC Center of Excellence in Minneapolis. The Center has been directly involved with several clinical studies including some drug trials. Although the clinic is not engaged in HD research on the molecular level, the HCMC is affiliated with the University of Minnesota where many groups are researching HD-related biological mechanisms. Several factors are considered when the clinic chooses which studies in which to participate: time involvement for participants and medical staff as well as whether the clinic has an appropriate patient population for the study. Research is mainly focused on relieving the symptoms of HD.
From Ms. Radtke’s experience with patients, families and caregivers are responsive to the idea of participating in clinical trials. Most HD patients are eager to take part in drug studies because receiving a new drug that is not yet FDA approved may alleviate symptoms. Patients are warned, however, that they may receive a placebo rather than the new drug itself.
HCMC researchers face several challenges when conducting clinical trials. Ms. Radtke tells HOPES that the greatest challenge in managing research at the Center of Excellence is time restraints. Some clinical trials require a significant time commitment from participants and all of the studies require time from the medical staff. Another challenge is keeping study participants involved. Although caregivers and family members are usually able to maintain their involvement in studies, it is difficult for HD patients to continue committing to studies as their disease progresses.
The HCMC Center of Excellence has been directly involved with several clinical studies and is currently collecting data for PHAROS, PREDICT-HD, and 2CARE.
PHAROS or the Prospective Huntington At Risk Observational Study monitors individuals who are at risk for HD but have not received genetic testing for the HD allele. By recording characteristics of these participants over time, researchers hope to better understand the progression of HD. At the HCMC Center of Excellence, Dr. Martha Nance is the Primary Investigator and Dr. Scott Bundlie is the independent rater who separately scores participants to ensure that the data is consistent. Ms. Radtke showed HOPES some of the PHAROS tests that are conducted at the HCMC Center of Excellence. Participants are monitored through performance on a verbal fluency test, the Stroop Test, the UHDRS, and questions about frequency and severity of behavioral symptoms.
The PREDICT-HD study is similar to PHAROS, but enrolled subjects are certain that they have the HD allele. The brains of both participants with HD and participants who are at-risk for HD are monitored and compared to better understand the neurological changes associated with HD.
2CARE is a phase III clinical trial investigating the therapeutic effectiveness of the anti-oxidant coenzyme Q10. Enrolled subjects have responded with enthusiasm to this drug trial. To learn more about HD and coenzyme-Q10 click here.
To learn more about clinical trials like the ones conducted at the HCMC and how drugs are developed click here.
Support and Outreach
The HCMC Center of Excellence works with the Minnesota Chapter of HDSA to offer support for HD families and caregivers. You can visit the HDSA website for the Minnesota Chapter by clicking here. There are monthly support group meetings facilitated by social worker Jessica Hancock. The groups are held in three different regions for the convenience of HD families: the Twin Cities, Northern Minnesota and Northwest Minnesota. In addition to giving HD families an encouraging environment, the support groups will occasionally have guests from the HD clinic to give presentations about health and medical topics. An annual Minnesota HD conference in September gives these groups an opportunity to meet with each other and hear from others who are closely involved with HD, like healthcare workers.
There are also collaborative efforts within the HD community to hold events to raise money for HD. Among these events are an annual Hoop-a-thon started by a local high school basketball player whose mother has HD. The Hoop-a-thon increases HD awareness and raises funds for HD medical research. Events such as the “Hits for Huntington’s” Golf Classic and “Hunt for the Cure” Charity Paintball Big Game also raise money for HD organizations and research.
Several HD families have taken the initiative to reach out to the community and encourage people to learn more about HD. One family whose inspiring story has increased awareness about HD within their community is the Johnson family. The family cares for Mr. Johnson’s childhood friend, Cory Daniels, who was diagnosed with juvenile HD at the age of eighteen, Cory was only given eight months to live when he was put in the care of the Johnsons. HOPES had the opportunity to speak with Mr. Johnson’s wife, Heather, who worked as a registered nurse at a long-term care facility where Cory lived. Heather reconnected the childhood friends who lost touch after high school The Johnsons were aware that community care is more beneficial for patients than institutionalization, so they worked hard to obtain certification so Cory could come to their house to live. Cory has been with the Johnsons for over four years despite physicians’ predictions that he would only live for another eight months. Through organizations like the HCMC center of Excellence and the Minnesota Chapter of HDSA as well as individuals like Cory and the Johnsons, the HD community in Minnesota hopes to continue providing support to one another and increase awareness about HD.
HOPES would like to thank the HDSA Center of Excellence at HCMC and Dr. Nance for allowing us to experience first-hand a clinic that provides quality care to HD patients. We would also like to extend our thanks to Research Coordinator Dawn Radtke for helping to arrange the visit and the Twin Cities Metro Area HD support group members for allowing HOPES to join their July meeting. To visit the website for the HCMC Center of Excellence click here.
More
Huntington’s disease (HD), an inherited neurodegenerative disorder, damages specific areas of the brain, resulting in movement difficulties as well as cognitive and behavioral changes. HD is often characterized by the motor symptoms that it causes.
Huntington’s disease (HD), an inherited neurodegenerative disorder, damages specific areas of the brain, resulting in movement difficulties as well as cognitive and behavioral changes. HD is often characterized by the motor symptoms that it causes. In fact, when HD was first discovered it was called Huntington’s chorea, as a reference to the uncontrollable, dance-like movement that is common among people with HD. Motor symptoms, though not always the first symptoms to appear, are often the reason that people with HD first see a doctor. Before genetic testing for the expanded CAG repeat within the Huntington gene became available, doctors could only make diagnoses according to motor symptoms. Even today, these symptoms are an important part of the criteria for clinical diagnosis; they generally define the age of onset of HD in an individual.
marker control range basic inhibition oxygen neurotrophic factor
What are the motor symptoms that occur with HD?
The progression of HD is different in every individual, but the following list contains most of the physical conditions that occur frequently in adult-onset HD. Keep in mind that not everyone with HD will experience all symptoms, and the progression from stage to stage is only a generalization. The time it takes to move from one stage to the next is also highly variable. It is important to note as well that juvenile HD exhibits motor symptoms that can be quite different from the adult form. (For more information on juvenile HD, click here).
Early stage
- Changes in coordination
- Some involuntary movement (such as irregular, sudden jerks of limbs)
- Fidgeting
- Restlessness, desire to move about
- Twitching, muscle spasms, tics
- Less control over handwriting
- Facial grimaces
- Difficulty with coordinated activities, such as driving
- Some rigidity
Middle Stage
- Dystonia (prolonged muscle contractions), often of the face, neck, and back
- More involuntary movements
- Trouble with balance and walking
- Chorea, twisting and writhing motions, jerks
- Staggering, swaying, disjointed gait (can seem like intoxication)
- Speech difficulties, including poor articulation, grunting, and abnormal speech patterns
- Problems swallowing
- Trouble with activities that require manual dexterity
- Slow voluntary movements, difficulty initiating movement
- Inability to control speed and force of movement
- Slow reaction time
- General weakness
Late Stage
- Rigidity
- Bradykinesia (difficulty initiating and continuing movements)
- Severe chorea (less common)
- Serious weight loss
- Inability to walk
- Inability to speak
- Swallowing problems, which create danger of choking
- Inability to care for oneself
Though HD is not fatal in and of itself, the conditions that it causes can eventually lead to death. One of the most serious concerns for people with late stage HD is loss of control of the throat muscles. This condition makes swallowing difficult, and ultimately, dangerous. Everyone’s body is constructed with two tubes that begin below the throat; one, the esophagus, leads to the stomach, and the other, the trachea, leads to the lungs. Usually, we have no trouble making sure that food passes through our esophagus and not into our trachea. We do this without thinking, and rarely does something go “down the wrong pipe.” For people with late stage HD, however, this process of sorting food and air often functions poorly. As a result, food can get caught in the trachea and lead to choking. If food gets caught in the lungs, it can lead to an infection known as aspiration pneumonia. Although most people recover from pneumonia, people with HD usually have compromised immune systems, and therefore are unlikely to recover from such a severe infection. (For more information on other potential complications of HD, click here.

If you would like to learn more about the specifics of motor symptoms and how they are used to diagnose and assess the stage of HD, click here).
What exactly is chorea?
Chorea is a disorder of the nervous system that occurs in multiple clinical conditions. In other words, it is not limited to HD, even though it is one of the classic symptoms associated with this particular disease. Chorea is characterized by spontaneous, uncontrollable, irregular movements, generally of the limbs and face. It can appear as unexpected jerks or twisting, writhing motions. These unpredictable movements contribute to poor balance, and the resulting walking difficulties lead to the staggering, swaying gait associated with HD. It is this irregular walking pattern that can make people with HD appear intoxicated, and also explains the root of the word chorea, which is the Greek word for dance. In the extreme, chorea can be a constant stream of violent movement. Severe choreic motions are known as ballismus.
Chorea occurs in 90% of people with HD, and increases over the first 10 years following onset. Although the specific motions of chorea can vary from one individual to the next, there are often consistent patterns within individuals. Chorea is usually present during waking hours, and cannot generally be suppressed. As HD progresses, chorea normally gives way to other movement difficulties, such as rigidity and bradykinesia.
To view a video clip showing chorea, please here. Footage is from Lake Maracaibo, Venezuela, courtesy of Dr. Nancy Wexler. HOPES would like to thank Julie Porter and the HD Foundation as well.
How can motor symptoms be treated?
Unfortunately, as there is no cure for HD, there is also no cure for the motor symptoms that accompany the disease. There are, however, drugs and supplements available that may lessen certain motor symptoms of HD. It is also possible to treat many of the behavioral symptoms, which can greatly improve quality of life. (For more information on drugs and supplements that are used to treat HD, click here, and for information about behavioral symptoms, click here). Under certain circumstances, there is a surgical procedure that can be performed, which involves making stereotactic lesions in a part of the brain called the thalamus. This procedure may alleviate motor symptoms, but it can only be performed when no cognitive decline is evident, and ultimately it does not halt the progression of the disease.
In addition to clinical treatments, there are other means of dealing with motor difficulties. One place to start is with health professionals: speech pathologists, physical therapists, and occupational therapists. Speech pathologists help with the mechanics of eating and drinking, as well as the loudness and articulation of speech. They can provide strategies for improving communication within the family and can also begin discussions about the use of a feeding tube, in the event that such a step becomes necessary. Exercise can be a very positive means of therapy, with physical, psychological, and emotional benefits. Physical therapists develop specialized exercise programs, usually to improve stretching and range of motion. They also advise people with HD about the use of walkers and wheelchairs. (For more information on exercise and HD, click here). Occupational therapists find ways to help people compensate for their inability to perform daily tasks, like eating and dressing. Often this involves adjusting the surrounding environment to better suit the needs of the individual with HD. Even small changes can make him or her feel more comfortable and capable, and thereby make his or her symptoms less problematic in daily life. (For suggestions on environmental adjustments, click here).
Motor symptoms can also be managed through lifestyle adjustments. Exercise, as previously mentioned, diet, and stress all affect overall health, and may contribute to the severity of symptoms. You should always consult your doctor before making any changes to your normal routine, but by clicking here, you can learn more about lifestyle adjustments that could potentially have positive effects.
What causes motor symptoms?
The reasons why HD causes motor symptoms are very complex and not entirely clear. However, researchers have learned a great deal about what may be at the root of the problem. In order to begin discussing why motor symptoms occur, we first have to look at how movement is organized in the brain. Motor control operates through two main pathways, which link the cortex (the outer part of the brain, responsible for sophisticated functions) with the basal ganglia (a grouping of cells found deep within the brain, responsible for more basic functions). These pathways are termed “direct” and “indirect.” Before continuing, you may want to take a moment to review these two pathways described here, in the Neurobiology of HD section.
After reviewing the basics of the direct and indirect motor pathways, we can examine this schematic diagram that combines the two (Figure 1). Notice that there is an additional pathway: nerve cells in the striatum also project, or link, onto a region of the basal ganglia called the substantia nigra (as well as the globus pallidus), which then projects directly back onto the striatum. Though it may seem odd to have a simple loop added to this system, we will see that this pathway, the striatonigral pathway, is very important to motor function.

In looking at the diagram, notice that along each projection arrow there is the name of a particular chemical, known as a neurotransmitter. Neurotransmitters are the means by which cells (and brain regions) communicate with each other. One cell, the presynaptic cell, releases a neurotransmitter and another cell, the postsynaptic cell, absorbs it. This chemical signal causes the postsynaptic cell to take some sort of action, such as releasing a neurotransmitter or actively not releasing one. Its response will then influence other cells farther down the line. This progression of cell-to-cell chemical communication is the nuts and bolts of the motor control pathways that we have been discussing.
You can see from the diagram that each motor pathway involves a complex combination of neurotransmitters. Let’s walk through the various pathways to get a clearer picture of how this all works. Remember though, it is the overall concept of the pathways that is important, not the names of each brain region and neurotransmitter.
The first step for all motor pathways is the cortex receiving sensory information from the outside world, via sight, touch, hearing, etc. It transmits this information to the striatum (part of the basal ganglia) in chemical form, using a common neurotransmitter called glutamate. Glutamate then causes the cells of the striatum to take action in the following ways:
The direct pathway: Nerve cells in the striatum project onto the internal part of the globus pallidus, using the neurotransmitters GABA and substance P. The cells of the globus pallidus then use GABA in their projections to the thalamus, a major relay and control center of the brain. The thalamus completes the loop back to the cortex using more neurotransmitters, sending its signals directly to the part of the cortex devoted to motor control, the motor cortex. The motor cortex responds to these signals (which originated in the basal ganglia, remember) by physically moving the body in the appropriate way.
The indirect pathway: Striatal cells (cells in the striatum) use GABA and enkephalin to project onto the outer part of the globus pallidus. Globus pallidus cells then project to the subthalamus using GABA, which in turn projects to the internal globus pallidus using glutamate. From there the pathway is the same as the direct pathway, progressing to the thalamus and then the motor cortex.
An important note: Certain neurotransmitters are termed “excitatory” and others “inhibitory.” Excitatory neurotransmitters cause an action to take place in another cell or part of the body. Inhibitory neurotransmitters prevent an action from occurring. All projections that come from the basal ganglia (including the striatum, globus pallidus, and substantia nigra) are inhibitory. We know that these cells are involved in controlling the movement of the body, so therefore the neurotransmitters from cells in the basal ganglia serve to prevent (or inhibit) movement. Imagine you are sitting at a desk, writing on a piece of paper. You are moving your hand and arm, but the rest of your body is still. In order to keep the rest of your body still, the cells in your basal ganglia are releasing inhibitory neurotransmitters constantly. In this state, cells are said to be operating at their baseline firing rate. “Baseline” refers to what is normal, because most of the time we want to prevent movement in at least some parts of our body, and “firing rate” refers to how frequently the neurotransmitters are released. Consider that while you are at the desk writing, you see that you have made a mistake. This visual sensory information reaches your cortex, and then is sent to your basal ganglia. The basal ganglia realize that you will need to tell your other arm to reach for an eraser. In order to stop inhibiting movement in that arm, the basal ganglia must adjust its release of inhibitory neurotransmitters. This modified signal is passed to the thalamus and then the motor cortex. Because the motor cortex is no longer inhibited as much, it can tell your other arm to reach for the eraser. When you have finished using that arm, neurotransmitter release returns to normal, to the baseline firing rate.
How does all this work in HD? Mutant huntingtin protein is expressed in all the cells of the body, but the most and earliest damage is seen in the basal ganglia, and the striatum in particular. The precise mechanism by which mutant huntingtin harms cells and causes them to behave differently is not clear. However, we know that mutant huntingtin causes serious problems with cell function and eventually leads to cell death. Here is where an understanding of motor pathways comes in handy. The early motor symptoms seen in HD are the result of damage to the striatum that impacts the indirect pathway (although both pathways are affected at the same time in juvenile HD). Damage from HD causes the striatum to release a weaker chemical signal, resulting in less inhibitory neurotransmitters, less inhibition of the motor cortex, and more movement. This movement is unintended, the result of a pathway error, and is therefore called “involuntary.” Involuntary movements include the fidgeting, tics, and chorea associated with early to middle stage HD. Later on in the disease the direct pathway becomes increasingly affected. In this case, the striatum still releases less inhibitory neurotransmitters, but in the direct pathway this action leads to more inhibition of the motor cortex and less movement. The result is rigidity of the body and bradykinesia, common to late stage HD. So, looking at how the direct and indirect motor pathways work and the motor symptoms we know to occur in HD, we can follow a logical route from damage in the striatum to actual symptoms. But what causes the neurotransmitter signals from the striatum to decrease in the first place? Let’s first take a look at the third motor pathway in the diagram.

The striatonigral pathway: Nerve cells in the striatum also project onto the substantia nigra, using GABA. The substantia nigra then responds with dopamine, projecting straight back onto the striatum. This dopamine signal influences both the direct and indirect pathways, but with different results, even though both pathways are responding to the same chemical signal. This is accomplished by having two different kinds of dopamine receptors on the post-synaptic cells in the striatum: D1 receptors link to the direct pathway and D2 receptors link to the indirect. Dopamine that goes to D1 receptors causes the striatum to release less inhibitory neurotransmitters, which ripples through the whole direct pathway and ultimately leads to inhibition of the motor cortex (preventing movement). Dopamine that goes to D2 receptors also causes the striatum to release less inhibitory neurotransmitters, but because of a different pathway progression, ultimately leads to less inhibition of the motor cortex (causing movement).

Researchers think that the answer to why HD causes the striatum to release a weaker chemical signal may be the striatonigral pathway and dopamine. As we have discussed, HD seems to over-stimulate the motor cortex via the indirect pathway and under-stimulate the motor cortex via the direct pathway. Interestingly, this pattern matches up with the influence of the striatonigral pathway on the other two pathways. When dopamine is released from the substantia nigra, it inhibits the striatum, causing it to release less inhibitory neurotransmitters. Let’s put these ideas together: if an excess of dopamine is released from the substantia nigra, the indirect pathway would over-stimulate the motor cortex and the direct pathway would under-stimulate it, just like in HD. You can see why researchers started to think that the striatonigral pathway and dopamine might be the key.

So what causes the substantia nigra to release more dopamine? For a potential answer we must trace the pathway back even further. Remember that as soon as the striatum receives a sensory message from the cortex, it sends a signal to the substantia nigra, via the neurotransmitter GABA, which then influences the substantia nigra’s release of dopamine. These two neurotransmitters go back and forth like a seesaw: more GABA means less dopamine and vice versa. Researchers have found that cells in the striatum that release GABA selectively degenerate due to damage from mutant huntingtin. GABA is an inhibitory neurotransmitter like all those in the basal ganglia. Therefore, if striatal cells are damaged and release less GABA, the substantia nigra is less inhibited and will release more dopamine. An increase in dopamine would inhibit the striatum, which is consistent with the pattern seen in HD.
It is important to note, however, that scientific studies have not been able to show conclusively that dopamine levels are increased in HD. Indeed, post-mortem studies of people with HD have shown elevated, depleted, and unchanged levels of dopamine in the brain. Additionally, the striatum uses GABA in its projections to both parts of the globus pallidus, not just the substantia nigra. Therefore, damage to the striatum from HD could lessen the release of GABA to the globus pallidus and thus the two main pathways directly, not just via the striatonigral pathway.
Nonetheless, many researchers are confident that dopamine is important to HD, even at endogenous, or natural, levels. Dopamine may in fact play an even more integral role in striatal cell damage, by causing the damage, not just influencing the pathway. One major question for researchers has been, why the striatum? Why is the basal ganglia harmed by mutant huntingtin, and not other cells? Recent studies suggest that the presence of dopamine is correlated with cell damage in HD. If this is the case, only cells in which dopamine was present would degenerate, and those with more dopamine would degenerate first. This theory would explain why cells in the striatum degenerate first. Charvin and others (2005) have shown that both dopamine and mutant huntingtin can activate a transcription factor known as c-jun. Transcription factors can influence a cell in many different ways; c-jun leads to programmed cell death, or apoptosis. When dopamine and mutant huntingtin are present together, the level of c-jun is greatly increased. The way that dopamine activates c-jun is as follows: dopamine can autooxidize, or in other words, spontaneously undergo a reaction that leads to reactive oxygen species (ROS). ROS are bad for the cell, and usually lead to cell damage. To prevent this damaged cell from hurting the rest of the body, the cell activates c-jun to start the process of programmed cell death (apoptosis). Therefore, the apoptosis of one cell is a good defense mechanism for the body. When mutant huntingtin is present, however, far too many cells induce apoptosis. Also, as we age, autooxidation of dopamine naturally increases. You can imagine that in someone with HD, more and more apoptosis due to dopamine combined with the presence of mutant huntingtin, could result in significant problems. This theory may therefore explain HD’s late age of onset. (For more information about the theory of oxidative stress and HD, click here).
Charvin and others proposed another role for dopamine in striatal cell damage. As previously mentioned, there are two kinds of dopamine receptors in the striatum: D1 for the direct pathway and D2 for the indirect. D2 receptors are more significantly implicated in HD. This makes sense, given that the indirect pathway is affected first. Charvin et al. suggest that D2 receptors are over-stimulated. Their theory also says that, as dopamine passes through the D2 receptors, it contributes to the formation of aggregates (or clumps) of the mutant huntingtin protein within the cell. Aggregates of mutant huntingtin are a common pathological marker in HD, meaning that they are present in cells affected by HD. It is unclear, however, what the function of these aggregates actually is. They may be harmful, helpful, or not have any effect on the cell at all. (For more information on protein aggregates, click here).
Scientific studies have consistently noted that dopamine receptors (D1 and D2) are depleted in HD. This may seem strange, as we have been suggesting that the presence of dopamine (or perhaps the excess of dopamine) is the reason why HD motor symptoms occur. Although the depletion of receptors is well known, the cause of the depletion is not. D2 receptors, for the indirect pathway, are depleted first, with more D1 receptors, for the direct pathway, disappearing as HD progresses. One possibility is that too much dopamine may be toxic to the receptors, thus killing them off. It may also be the case that cells try to protect themselves from an excess of dopamine, or its toxic influence in the presence of mutant huntingtin, by actively losing receptors. Another possibility is related to brain-derived neurotrophic factor (BDNF). BDNF is a chemical that protects cells in the brain, and its function has been shown to be impaired in HD. The loss of BDNF could make it much easier for receptors to be damaged, as well as allowing for the mutant huntingtin/dopamine synergistic damage to occur in the first place. (For more information on BDNF, click here). It is also possible that mutant huntingtin harms receptors directly. Regardless of the specific cause of receptor depletion, much damage from dopamine can occur by the time depletion becomes significant. Additionally, if the striatum is absorbing less dopamine, an increased release of dopamine could be triggered in the substantia nigra. A reduced number of receptors can also lead to greater sensitivity of the remaining receptors, ultimately resulting in more dopamine absorption and damage. As you can see, cell-to-cell communication is very complex and intricate. Though this fact makes it difficult to determine just how HD affects the brain, it does give researchers many ideas about what to look at next, as well as offer many possibilities for treatments.
So what does all this mean for HD treatments? Currently in the U.S. there are few medications that are prescribed to treat motor symptoms of HD, and none that are particularly aimed at chorea. However, experimental drugs that deplete dopamine have been reported to have positive effects on motor symptoms. The best-studied drug, tetrabenazine, should soon be available in the U.S. and will be discussed in detail in the chapter linked to below. As we learn more and more about the cause of HD damage in the brain, we can develop new treatments that are aimed at specific mechanisms. Future medications may target ROS production, dopamine absorption through D2 receptors, or initiation of the c-jun pathway, to name a few. These new kinds of treatments will hopefully prove to be more effective than current options, impacting the progression of HD in a meaningful way.
Tetrabenazine
Click here for an article about Tetrabenazine
For further reading
- Bates, G., Harper, P., & Jones, L. Huntington’s Disease. New York: Oxford University Press, 2002. pp. 28-37, 276-281.
This book is a thorough review of current knowledge about HD, but is very scientifically-oriented.
- Canals, J.M., et al. “Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease.” 2004.Journal of Neuroscience. 24(35): 7727-7739.
An article about BDNF and HD.
- Charvin, D. “Unraveling a role for dopamine in Huntington’s disease: the dual role of reactive oxygen species and and D2 receptor stimulation.” 2005. PNAS? 102(34): 12218-12223.
This article presents the possible mechanisms for how dopamine may damage striatal cells.
- Dr. Joseph F. Smith Medical Library. “Huntington’s disease.” http://www.chclibrary .org/micromed/00051720.html
A description of motor symptoms and alternative treatments for HD, such as occupational, speech, and physical therapies.
- Emedicine.com. “Huntington disease dementia.” http://www.emedicine.com/me d/topic3111.htm
Brief review of motor symptoms associated with HD.
- Gazzaniga, M.S., Irvy, R.B., & Mangun, G.R. Cognitive Neuroscience: The Biology of the Mind. New York: W.W. Norton & Company, 2002. pp. 488-492.
This is a textbook covering many topics in neurobiology. It is rather technical.
- HDNY at Columbia University. “Speech pathology”: http://www.hdny.org/speech.html, “Social implications of motor disorders”: http://www.hdny.org/problems.htm, “Environmental adjustments”: http://www.hdny.org/rehab.html
The HDNY website is very helpful as a general resource, even outside the NY area. These pages are particularly relevant to motor symptoms.
- Health-cares.net. “What is Huntington’s chorea?”: http://neurol ogy.health-cares.net/huntingtons-chorea.php, “What causes chorea?”: http://neurology.he alth-cares.net/chorea-causes.php
This website discusses HD and other forms of chorea.
- Hickey, M.A., et al. “The role of dopamine in motor systems in the R6/2 transgenic mouse model of Huntington’s disease.” 2002. Journal of Neurochemistry. 81: 46-59.
A good study of dopamine and HD in a mouse model.
- International Huntington Association. “Huntington’s disease.” http://www.huntington-assoc .com/huntin.htm
A summary of the progression of HD, in terms of motor, cognitive, and behavioral symptoms.
- Jakel, R.J., & Maragos, W.F. “Neuronal cell death in Huntington’s disease: a potential role for dopamine.” 2000. Trends in Neuroscience, 23: 239-245.
This is a good article that reviews the potential mechanisms for cell damage as a result of HD.
- Nieuwenhuys, R., Voogd, J., & van Huijzen, C. The Human Central Nervous System: a Synopsis and Atlas. New York: Springer-Verlag, 1981. pp. 169-173.
A highly technical book that details neuroanatomy.
- Petersen, A., et al. “Mice transgenic for exon1 of the Huntington’s disease gene display reduced striatal sensitivity to neurotoxicity induced by dopamine and 6-hydroxydopamine.” 2001. European Journal of Neuroscience. 14:1425-1435.
This is a rather complex article that discusses the potential for dopamine toxicity in striatal cells.
- Pineda, J.R., et al. “Brain-derived neurotrophic factor modulates dopaminergic deficits in a mouse model of Huntington’s disease.” 2005. Journal of Neurochemistry. 93: 1057-1068.
More on BDNF.
- Reynolds, D., et al. “Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington’s disease.” 1998. Journal of Neuroscience. 18(23): 10116-10127.
This article is one of the earlier articles to show that dopamine is important to cell damage in HD.
- UCLA Medical Center. “How is Huntington’s disease treated?” http://neurosurgery.ucla.edu/Diagnoses/Movement/MovementDis_2.html
A brief overview of medical and surgical treatment options, from the neurosurgery department at UCLA.
- We Move. “Medical management of Huntington’s disease.” http://www.wemove.org/hd/hd_tr e_mm.html
A brief discussion of available medical treatments for HD and their potential consequences.
C. Tobin 6-29-06
More
Dementia refers to neurodegeneration that results in loss of mental abilities. Neurodegeneration is the loss of mental abilities that can be caused by brain damage and/or neuron death. For this reason, dementia is common in neurodegenerative disorders such as Alzheimer’s Disease. While Huntington’s Disease (HD) is commonly thought of as a motor disorder, cognitive symptoms can be present which can progress to dementia. To learn more about some of these cognitive symptoms, click here. Interestingly, many cognitive symptoms appear in HD patients before motor deficits appear.
Although a formal clinical diagnosis of HD depends on unequivocal signs of motor impairment, recent research has shown the importance of neuropsychological analysis and the evaluation of dementia in determining the condition of HD patients. There are several tests that clinicians administer to evaluate a patient’s cognitive abilities and degree of dementia. For physicians, it is important for these tests to recognize the subtle differences between different neurodegenerative diseases, particularly HD, Alzheimer’s disease and Parkinson’s Disease, which show similar cognitive symptoms.
Testing for Dementia
When dementia is suspected in patients, physicians will administer tests before giving a formal diagnosis. For patients, whether they are at risk for HD or other diseases associated with dementia, the Mini-Mental State Examination (MMSE) is the most common test that is administered. The MMSE is convenient to administer to patients because it is relatively short, but can still help determine whether a patient’s cognitive functions are declining.
In addition to the MMSE, physicians also use other neuropsychological tests that usually involve several mental tasks that require the use of different areas of the brain. For example, the striatum, the brain area most affected in HD, has been implicated in sequence and procedure learning. Since the neuropsychological tasks test different regions of the brain, physicians can use the results to determine what regions of the brain have been affected. When patients are tested for dementia using these tests, their performance is compared to that of healthy individuals on the same tasks. Researchers are still determining which neuropsychological tests are most suitable for evaluating certain regions of the brain.
How are dementias classified?
After a patient is diagnosed with dementia, it is important to determine the kind of dementia that is present. Dementias are often classified by the region of the brain that is affected. One of the main classifications divides dementias into two main groups: cortical and sub-cortical based on the area of the brain where degeneration occurs. The cortical region consists of the cerebral cortex while the sub-cortical region is comprised of the other structures of the brain including the thalamus, hypothalamus, cerebellum and brain stem (See Figure 1). To learn more about the brain click here for the HOPES Brain Tutorial. Whether cortical and subcortical dementias should be considered separately is still controversial among researchers and physicians. In general, studies have shown that some differences do exist, but there is disagreement on the degree to which the two dementias differ.
Figure 1: The brain can be divided into the cortical and subcortical regions. The cortical region consists of the cerebral cortex while the sub-cortical region consists of the thalamus, hypothalamus, cerebellum and brain stem.
To more clearly define the two types of dementia, researchers have studied whether their effects on memory differ. Alzheimer’s patients are often used as a model for cortical dementia because patients with this disease have large amounts of degeneration in the cerebral cortex. Clinical studies of Alzheimer’s patients have shown that cortical dementias have difficulty performing tasks that require semantic memory. Semantic memory is what we use to store facts without respect to the setting where we learned the facts (See Figure 1). To evaluate this type of memory, patients are asked to perform tasks such as matching pictures and generating definitions of words. To a lesser degree, cortical dementia can also affect episodic memory, which is used to remember experiences and the setting in which facts are learned. For example, after a boy bumps his head in a bike accident, semantic memory would enable him to remember that wearing a helmet is important when riding bikes, while episodic memory would enable him to recollect the specific time when the accident occurred.
Sub-cortical dementias have a slightly different effect on memory than cortical dementias in that they have a smaller effect on semantic memory. HD and Parkinson’s disease are considered sub-cortical dementias. In HD, patients instead find it challenging to accomplish cognitive tasks that require retrieval and synthesis of known facts, such as forming abstractions. Unlike patients with Alzheimer’s, however, those with HD can accomplish tasks that require semantic memory under the right conditions. For example, one study tested patients on a category fluency task in which patients were asked name as many items as possible from certain categories (e.g. foods, animals, plants) within an allotted time period. Under what scientists call “cued” conditions in which patients were given hints or clues that help with the task, HD and Parkinson’s patients improved their scores. Alzheimer’s patients, however, did not perform better under “cued” conditions. This suggests that patients with HD or other types of sub-cortical dementia have not experienced degradation of semantic memory per se, but instead have difficulty retrieving facts from their memory.
Sub-cortical dementias like HD do not affect memory following a time-dependent gradient. Memories and knowledge obtained recently are not more susceptible to degeneration than those from the distant past, as is the case in Alzheimer’s Disease. To learn more about the effects of HD on memory click here.
Figure 2: Our brains use different types of memory. Cortical dementia is thought to affect semantic memory to a greater degree than episodic memory. Sub-cortical dementia, as in HD, is thought to affect semantic memory to a lesser degree than cortical dementias.
Although memory is one of the leading areas of interest in the study of cortical and subcortical dementias, other differences between the two dementias exist. Sub-cortical dementias almost always result in motor disorders. Chorea in HD patients and tremors in Parkinson’s tremors are examples of motor impairment that accompany sub-cortical dementias. In terms of other cognitive effects, differences between dementias are still being studied.
Criticisms of the Dementia Differentiation
Some researchers and physicians consider the differentiation between cortical and sub-cortical dementia important for patient diagnosis, but others remain skeptical that a significant difference exists. The major criticism of the studies that show variation between cortical and sub-cortical dementias is that there is pathological overlap between the sample groups that are used to model the two categories. These studies often assume that Alzheimer’s patients mostly have cortical dementia and HD or Parkinson’s patients preferentially exhibit subcortical dementia. Necropsies have shown, however, that the brains of both Alzheimer’s and HD patients exhibit a certain degree of both categories of dementia.
If in fact both cortical and subcortical dementia occur in Alzheimer’s, HD, and Parkinson’s patients, then these studies may be problematic. As a result, physicians are still trying to learn more about the differences between the pathologies of the diseases in hopes of finding a more reliable way of differentiating dementias. The ability to differentiate dementias may lead researchers and physicians to better diagnose and treat neurodegenerative diseases.
Further Reading
- Langbehn, Douglas R. et al. “Predictors of diagnosis in Huntington disease.” Neurology. 2007; 68: 1710-1717.
Researchers and The Huntington Study Group performed a longitudinal study to identify early clinical symptoms that arise in HD patients. Findings showed that psychological performance can be used in diagnosis along with motor impairment. There is a thorough discussion of clinical evaluation of neurological performance in HD patients and the types of tests administered to patients.
- Rosser, Anne and John R. Hodges. “Initial letter and semantic category fluency in Alzheimer’s disease, Huntington’s disease and progressive supranuclear palsy.” Journal of Neurology, Neurosurgery and Psychiatry 1994; 57: 1389-1394.
This study examined symptoms related to semantic and episodic memory in three different neurodegenerative diseases. Several neuropsychological tests were administered to patients and the results suggested that semantic memory is more heavily influenced in cortical dementias like Alzheimer’s disease.
- Sadek, Joseph R. et al. “Retrograde Amnesia in Dementia: Comparison of HIV-Associated Dementia, Alzheimer’s Disease, and Huntington’s Disease.” Neuropsychology, 2004; 18.4: 692-699.
This study tested how three different types of dementia affect memory. The findings show that overall dementia is equally severe in all three types, but memory impairment differs. Time-dependent memory loss was not found in HD patients and HD patients were able to improve on memory tasks under “cued” conditions. The authors discuss their findings in the context of the debate on how cortical and subcortical dementias differ.
- Wedderburn, C et al. “The utility of the Cambridge Behavioural Inventory in neurodegenerative disease.” Journal of Neurology, Neurosurgery, and Psychiatry. 2008; 79: 500-503.
A review of a new test that is used to evaluate the mental condition of patients with neurodegenerative diseases. It includes helpful information about cognitive and psychological symptoms in HD, Parkinson’s and Alzheimer’s patients and how these symptoms differ between the diseases.
Additional Resources:
- Ho AK, Sahakian BJ, Brown RG, Barker RA, Hodges JR, Ane MN, Snowden J, Thompson J, Esmonde T, Gentry R, Moore JW, Bodner T (2003) “Profile of cognitive progression in early Huntington’s disease.” Neurology 61:1702-1706.
- Kirkwood SC, Siemers E, Hodes ME, Conneally PM, Christian JC, Foroud T (2000) “Subtle changes among presymptomatic carriers of the Huntington’s disease gene.” J Neurol Neurosurg Psychiatry 69:773-779.
- Lawrence A, Hodges J, Rosser A, Kershaw A, French-Constant C, Rubinsztein D, Robbins T, BJ S (1998) “Evidence for specific cognitive deficits in preclinical Huntington’s disease.” Brain Pathol 121:1329-1341.
- Lemiere J, Decruyenaere M, Evers-Kiebooms G, Vandenbussche E, Dom R (2004) “Cognitive changes in patients with Huntington’s disease (HD) and asymptomatic carriers of the HD mutation—a longitudinal follow-up study.” J Neurol 251:935-942.
- Meade, Catherine E. “Diagnosing Dementia: mental status testing and beyond.” Australian Prescriber, 2005 (28): 11-13.
- Savla, Gauri Nayak and Barton W. Palmer. “Neuropsychology in Alzheimer’s disease and other dementia research.” Current Opinions in Psychiatry, 2005 (18): 621-627.
– T. Wang, 5/17/09
More
Cognitive-Symptoms-of-HD
Huntington’s Disease (HD), an inherited neurodegenerative disorder, damages specific areas of the brain, resulting in movement difficulties as well as cognitive and behavioral changes. The term “cognitive” refers to tasks of the brain that involve knowing, thinking, remembering, organizing, and judging. Certain changes in cognitive abilities are characteristic of HD and can significantly impact the lives of individuals with the disease. For example, cognitive changes may affect the ability of a person with HD to work, manage a household or properly care for him or herself
range folding marker basic control
What are some of the cognitive abilities that may be impaired by HD?
Communication
Communication is a complex process, requiring the cognitive ability to express and understand as well as physical abilities such as muscle control and breathing. Typically, neural degeneration, resulting from HD, begins in the core of the brain at the caudate nucleus and may spread to areas on the left and right side of the brain, such as the control centers for cognitive function, speech and language. Thus, communication problems tend to become more prominent as the disease progresses.
Throughout the course of the disease, communication problems vary in nature and severity. However, variations in the nature and severity of communication problems also occur from person to person. While one individual may have difficulty initiating conversation, another may have very little difficulty initiating, but severe difficulty word-finding (an aspect of memory recall). Although there are a number of communication problems that may arise for people with HD, the most common communication difficulties are four: speaking clearly, initiating conversation, organizing what is to be said and understanding what is being said.
As HD damages neurons in the caudate, proper regulation of motor information that tells the body how to move specific muscles at precise times may be impaired. The caudate’s inability to regulate motor information can result in slurred speech and stuttering as well as the uncontrolled bodily movements, often referred to as chorea.
The ability to initiate conversation or activities is a very complex brain function. Damage to the caudate affects the brain’s ability to regulate the sequence and amount of information being transmitted, which may result in difficulty starting and stopping communication. The inability to initiate conversation may also be the result of word- finding difficulty. As neurons in the caudate die, the intact neurons have more difficulty sending information along the neural “circuit.” For example, it may take longer than expected for a person with HD to answer a question because it may be more difficult to find the right word. While word-finding is often impaired, knowledge of vocabulary is retained.
As the caudate and its connections with other areas of the brain deteriorate, some kinds of information may not reach the frontal lobes. Without the frontal lobes to sequence and prioritize outgoing information, the speech of a person with HD may become garbled or seemingly illogical. Damage to the caudate, resulting in impaired access to the frontal lobes may also make it difficult for an HD-affected person to understand what is being said; however, the ability to understand usually remains intact, even in the later stages of the disease. For example, each word of a sentence may be understood, but the frontal lobes and caudate may not be able to organize them properly, possibly resulting in miscommunication. This inability to organize incoming information can also contribute to a slowed response time, even if comprehension remains normal.
If you are interested in reading about strategies and tools that may improve communication for and with a person who has HD, click here.
Memory
An individual suffering from the cognitive symptoms of HD may have memory difficulties. It is important to note that the memory problems that can occur in people with HD are different from the memory difficulties that can occur in people with Alzheimer’s Disease. Whereas people with Alzheimer’s Disease may get lost in familiar places or forget the names of familiar people, individuals with HD will know and recognize people as well as places.
(Table adapted from Paulsen Understanding Behavior in Huntington’s Disease)
Throughout the course of HD, there are two primary memory difficulties that result from cognitive impairment: learning new information and recalling stored information. The impaired ability to learn new information may be the result of damaged neural connections between the frontal lobes and the caudate in the brain. Without efficient use of the frontal lobes, the brain cannot effectively organize and sequence the information to be learned. For example, learning a new phone number may be very difficult for an individual with HD because the brain may not organize or group the numbers together in a way that is easy to remember. For example, the series of numbers “3456978” is much more difficult to remember than “3-4-5-69-78.” When information is not organized in an efficient manner, retaining and recalling the learned information is very difficult.

Recalling stored information is the other primary memory problem for people with HD. For example, a hypothetical person, Silvia, knows what she had for dinner last night, but may not respond very quickly when asked. However, if you ask her whether she had pizza or chicken, she’ll be able to correctly identify which of the two choices she had for dinner. The neurodegenerative nature of HD disrupts the brain’s search mechanism, which makes recalling stored information more difficult, although the memory likely remains intact and can often be recalled through cues or recognition. Also, the person suffering from memory difficulties usually maintains the ability to understand and comprehend information.
Although most memories remain intact, motor memories are often impaired. Motor memories, such as driving a car or tying shoes, are considered implicit or “unconscious” memory. The impairment of these motor memories means that a person has to rely on “conscious memory” to perform these tasks, which requires more concentration. Since these simple, once automatic, tasks may require more concentration, people with HD often have difficulty multi-tasking or dividing their attention. For example, an individual suffering from memory problems due to HD may have difficulty making dinner while listening to the radio.

Recognition memory: stored information can be recalled through a cue. For example, an individual may not remember what time his haircut appointment is scheduled for, but when asked, “are you getting your haircut at 1:00 or 2:00?” he remembers that the appointment is at 2:00
Long-term memory: stores an unlimited amount of rehearsed information; each memory can be stored for a long period of time
Language comprehension: ability to understand the meaning of words as well as how they are organized in order to understand what is being said
Memory retrieval: recalling stored information
Verbal fluency: ability to use and organize words in order to clearly express thoughts, feelings and ideas
Word finding: recalling and using the proper word to communicate
Executive Functions
Critical to our ability to function effectively at home or work, the “executive functions” include prioritizing, problem solving, judgment, abstract thinking, controlling emotions and awareness of self and others. The frontal lobes, often referred to as the “boss” of the brain, are in charge of the executive functions. The part of the brain responsible for regulation information being sent to the frontal lobes is the caudate nucleus. When HD destroys neurons in the caudate nucleus, a person with HD may have difficulty efficiently performing tasks that were previously simple, such as running errands.
Many of the executive functions that may be impaired in individuals with HD fall into one of three categories: awareness, organization and regulation.
Awareness
Commonly, denial is used to describe the inability to accept the reality of a distressing circumstance. HD sufferers may deny having HD or be unable to recognize their disabilities. However, this denial is not under the individual’s control, so a lack of awareness or “unawareness” may be a more accurate word for people with HD.
Due to HD, circuits connecting the caudate nucleus, frontal, and parietal lobes may incur damage, resulting in a lack of self-awareness. People with HD may be unable to recognize disabilities or evaluate their own behavior. The inability to evaluate one’s own performance may cause sufferers to be unaware of mistakes that are evident to others. Damage to these neural connections may also impair the ability to experience a range of subtle emotions and see another’s point of view, possibly making social and personal relationships more difficult.
Unawareness often plays a role in seemingly irrational behaviors. For example, a person may become upset if he or she is not allowed to go back to work or live independently, because of the unawareness of failing capabilities. However, a person may be willing to talk about his or her capabilities, but still be unable to acknowledge that failing capabilities are the result of HD. Unawareness, a behavioral as well as a cognitive symptom, is generally accepted as an untreatable component of HD. To learn about the behavioral symptoms of HD, click here.
Organization
Since HD damages the caudate nucleus, many aspects of behavioral and intellectual functioning can be affected. The task of the caudate is to organize, regulate and prioritize information transmitted from many areas of the brain to the frontal lobes. If the information reaching the frontal lobes is not organized as a result of HD, the individual with HD may experience difficulty organizing his/her thoughts and activities as well.
In order to plan and prioritize efficiently, our brain must be able to organize activities in a logical order, evaluate all of the steps involved in accomplishing a task, and even think about one particular task while performing another. As a result of the damaged caudate, the brain of an individual with HD may not be capable of performing in such a manner. For example, a person without HD may spend one hour on a trip to the grocery store and the bank. However, it may take a person with HD two or three hours to accomplish this same task. While at the grocery store he or she may have to look for each item in the order of the list, possibly failing to get two items from the same aisle because they appeared in different places on the list.
A diminished ability to make decisions may also become a problem as a result of the brain’s failing organizational capabilities. If asked the question, “What would you like to have for dinner?” it may take a while for the brain of a person with HD to organize the words into an understandable question, retrieve the memories of past dinner items, process the feelings regarding each dinner item, and organize the words into a logical response. This difficulty may also be due to memory impairments resulting from HD. Thus, the process of decision making is drastically simplified if a person with HD is given choices, which allows the brain to recognize memories rather than retrieve them. Shorter sentences may also aid in the decision making process, as they contain fewer words for the brain to organize.
Another function affected by the impairment of the brain’s organizational capacity is attention. While simple attention, the ability to focus on one activity, often remains intact, sustained attention as well as divided attention may become impaired. As a result of memory impairments, “unconscious” tasks that were once automatic may require intense concentration. This makes dividing one’s attention very difficult. For example, it may be difficult for a person with HD to walk while carrying on a conversation. With the loss of motor memories, he or she may have to consciously think about each step forward, making conversation difficult. To read more about memory impairment as a cognitive symptom of HD, click here.
Regulation
The caudate nucleus serves primarily as a regulator and organizer. It controls the order and amount of information traveling from particular areas of the brain to the frontal lobes. As HD progressively destroys the caudate, it may become difficult for individuals with HD to initiate, maintain, and/or stop behaviors or thoughts.
As mentioned above, the ability to initiate activities or conversation is a complex brain function. Damage to the caudate disrupts the brain’s ability to regulate the sequence and amount of information being transmitted, which may result in difficulty starting and stopping communication or activities. The diminished regulatory abilities of the caudate may also result in the inability to maintain an activity or conversation. However, this may be due to the impairment of sustained attention as well. For example, an individual with HD may be able to begin folding laundry but quickly become unable to focus on the task at hand due to distractions. If the radio is playing in the room, the individual may focus his or her attention on the music and be unable to re-initiate and complete the task of folding laundry.
Another possible result of the caudate’s inability to regulate the amount of information traveling to the frontal lobes is a lack of emotional control. A person with HD may over-express a feeling of slight frustration or irritation in the form of a temper tantrum or aggressive behavior. Although the emotion itself is often a legitimate response to something in the individual’s environment, the caudate cannot regulate the proper amount to be expressed. To read about the behavioral symptoms of HD, including frustration, apathy and others, click here.
Visual Spatial Ability
Visual spatial ability is the ability to perceive one’s body position in the environment. An individual’s perception of his or her body position is useful for judgment of where he or she is in relation to walls or how close his or her hand is to a burner on the stove. Impaired visual spatial ability is often evident even in the early stages of HD. Most commonly, the individual suffering from cognitive symptoms of HD is aware of his or her visual spatial impairment.
For example, due to a diminished visual spatial ability, it may be more difficult for a person with HD to read a map or follow directions, since most directions are given using spatial cues, such as “east” and “west” or distances measured in miles. However, a person suffering from this cognitive symptom of HD may be able to follow directions if they are given using geographic markers, such as: “Go straight on Campus Drive until you reach a stoplight. Turn left and go past the Pet Store. The Post Office will be on the left side of the street with a flag pole in front.” For an individual suffering from visual spatial impairment, directions using “left” and “right” or geographic markers are easier to follow because they do not require the individual to orient his or her body in a particular direction. Regardless of which direction a person is facing, “left” is one way and “right” is the other. However, depending on the orientation of one’s body, “east” may be behind, to the right, to the left or in front of him.
Reading difficulties may also be the result of visual spatial impairment; however, the inability to maintain attention may be a contributing factor as well. For information about attention impairments as a cognitive symptom of HD, click here.
What causes the cognitive symptoms?
As a neurodegenerative disease, HD damages many neurons and neural connections within the brain, potentially causing cognitive impairment. Most of the damage occurs in the caudate nucleus and putamen, which are structures of the basal ganglia. To learn more about these brain structures, click here. The primary function of the caudate is the regulation and organization of information being transmitted to the frontal lobes from other areas of the brain. The frontal lobes are responsible for many important tasks, some of which are:
- Organizing
- Prioritizing
- Controlling impulses
- Self-awareness
- Initiating and ending activities
Thus, damage to the many connections between the caudate and frontal lobes can significantly impair cognitive abilities, such as reasoning, planning, attention, memory, and learning. To read about neurons and neural connections, click here.
The neurodegenerative changes that occur within the brain of a person who has HD are generally the primary cause of the cognitive symptoms of HD, as well as behavioral changes and movement difficulties. An individual suffering from the cognitive symptoms of HD may have difficulty effectively prioritizing his or her daily activities, initiating conversation or activities, recalling memories or making decisions. However, it is important to remember that the cognitive as well as behavioral and physical symptoms of HD vary from person to person. To learn about the behavioral symptoms of HD, click here.
How do the cognitive symptoms change as HD progresses?
As a general rule cognitive impairments tend to increase in severity as HD runs its course. However, only a few longitudinal studies have been done on the cognitive symptoms of HD, and thus, research has not determined whether the severity of a cognitive symptom can be used as a marker for the underlying progression of the disease.
Although the symptoms of HD vary significantly from person to person, there are some general trends among individuals. Speed of mental processing, organization, and initiation are commonly impaired early in HD and may worsen during the intermediate stages. While individuals with HD are often unable to speak or express their views in the later stages of HD, some cognitive abilities, such as the ability to understand incoming information, may remain relatively intact.
Do the cognitive symptoms of HD vary from person to person?
The expression of HD varies significantly from person to person. Although HD is a progressive disease for affected individuals, there is considerable variation in the type and severity of symptoms a person with HD may experience. Some individuals may experience a number of cognitive and behavioral symptoms and fewer physical symptoms, whereas others may suffer more from physical symptoms, such as chorea. The variation in severity means that while some of the cognitive symptoms may be quite pronounced for one person, those particular symptoms may be much less evident in another.
Due to the variation in the type and severity of cognitive symptoms, it may not be useful to use them as an indicator for the onset of HD in an at-risk individual or to diagnose the individual with the disease. Many of the early cognitive symptoms of HD, such as forgetfulness, lack of initiation or fumbling are also fairly common among individuals who are not at risk for HD. “Symptom watching” by individuals at risk for HD may result in a misinterpretation of these thoughts, actions or behaviors as HD. Genetic counselors may be contacted if symptom-watching or anxiety due to being at-risk for HD begins to interfere with one’s ability to function effectively.
Are there treatments available for the cognitive symptoms of HD?
At the time of this writing (April 2003), there is no cure for the cognitive symptoms of HD or the disease itself. The cognitive symptoms of HD are due to the damage of neurons and neural connections in the brain, which at this time are considered irreversible. However, scientists and researchers continue to investigate the brain’s ability to produce new neurons as well as its ability to form new connections between neurons. For more information about the brain’s natural reparatory ability, click here.
Fortunately, there are a number of strategies for coping with and enhancing cognitive abilities impaired by HD. For example, maintaining a calm, predictable environment and establishing routines can improve organization and planning as well as minimize the occurrence of emotional outbursts. A predictable, routine environment enables a person suffering from the cognitive symptoms of HD to organize daily tasks and adhere to that schedule, resulting in fewer organizational or planning problems. There are a number of resources that provide strategies for improving the cognitive symptoms of HD. If you are interested in learning more about these strategies, click here.
Although there are strategies and treatments that can improve the physical, behavioral and cognitive symptoms of HD, there are currently no treatments available that slow down the progression of HD. However, research continues with the growing hope of discovering effective treatments as well as a cure for HD. For more information on potential treatments for HD, click here.
For further reading
- American Speech-Language-Hearing Association. Huntington’s Disease. http://www.asha.org/speech/disabilities/Huntington-Disease.cfm
Much like a brochure, this resource explains many of the communication and swallowing problems that may occur as a result of HD. It also lists ways in which a speech-language pathologist can help minimize difficulties.
- Australian Huntington’s Disease Association. Information – Huntington’s Disease https://www.huntingtonswa.org.au/information/
This website provides basic information about HD as well as support for individuals with HD and their families.
- Huntington’s Disease Association. Fact Sheet 10. Behavioral Problems in Huntington’s Disease. https://docs.google.com/a/hda.org.uk/uc?id=0B7QMCpPHjjhuZF9wZzZkQl8yUkE&export=download
This clearly-written Fact Sheet is just one of the many informative and helpful Fact Sheets available at this site. The list of Fact Sheets can be found here.
- Paulsen, J. “Understanding Behavior in Huntington’s Disease” (2nd ed.). Huntington’s Disease Society of America, Inc, 1999.
This is an easy to read and extremely informative resource with the purpose of providing practical and helpful information about the behavioral as well as cognitive symptoms of HD.
- Rosenblatt, A., Ranen, G., Nance, M. & Paulsen, J. “A Physician’s Guide to the Management of Huntington’s Disease.” (2nd ed.) Huntington’s Disease Society of America, Inc., 1999.
This is a guide specifically for physicians. It explains the symptoms of Huntington’s Disease and methods for providing optimal management of the symptoms.
K.Hammond 3-29-03; recorded by B. Tatum 8/21/12
Podcast: Play in new window | Download
More
Huntington’s disease presents unique psychosocial issues due to its late onset and hereditary nature. One of the major issues of course is stress, which can come from many sources and has many effects (for general discussion of stress and HD, click here). A major source of psychosocial stress associated with HD comes from predictive testing which became available in the United States in 1993.
Extensive research has focused on the person undergoing predictive testing, with a good number of studies reporting that the tested person’s benefit from the knowledge of their genetic status outweighed their post-test psychosocial distress. However, less research has focused on the psychological impact that predictive testing may have on those at risk for HD and their partners, family and friends. This research is important because HD affects many more people than just the person who has it. Moreover, the hereditary nature of the disease can also lead to difficult questions about reproduction and about the possibility of other family members having the disease.
Fortunately, researchers are now focusing more of their attention on predictive testing and its effects on the couple relationship. In the remainder of this section, we review their key findings to date.
What percentage of couples looks favorably upon predictive testing? And what motivations drive their decisions?
In a 1989 study in Belgium, where HD predictive testing has been available since 1987, Evers-Kiebooms found that a moderate majority of people at-risk for HD and their non-carrier partners looked positively on predictive testing. Out of 349 study subjects, 66% of the at-risk adults and 74% of their partners wanted testing for the at-risk individual. The difference between these percentages can partly be explained by the difference in motivations between the at-risk person and his/her partner in approving the predictive testing. When asked why they approved, at-risk adults tended to cite worries about their futures, while their partners tended to cite worries about current and/or future children. A reason that some couples decided not to undergo predictive testing was concern about the effects of the testing on their relationship; this concern was more often a major consideration for non-carrier partners than for the at-risk adults. (For specifics on the process of HD predictive testing, please click here.
Is there a theory on how predictive testing affects couple relationships?
Yes, a perspective called family systems theory, developed over the past few decades, has proven particularly useful in genetic counseling. This theory is especially relevant to the genetic counseling of couple relationships because its central focus is on the family rather than the individual. The family systems theory describes human behavior as a consequence of family relationship patterns, rather than individual psychology. Consequently, family systems theory can help explain the effects of predictive testing on couple relationships by analyzing how family relationship patterns can influence post-test behavior.
In a 2004 study by Richards and Williams, 43 couples were divided into two groups: those that chose to undergo predictive testing and those that chose not to. Couples in both groups answered the same questionnaire before predictive testing, then 6 months later (3 months after those tested received their test results), and again 24 months after the first questionnaire. The questionnaire consisted of 32 Dyadic Adjustment Scale questions that measured couple relationship functioning, known as a “couple score.” Those couples that received higher couple scores frequently interacted and communicated with each other, rarely disagreed with each other on significant marital issues, and settled disagreements in a way that was satisfying to both partners.
The major finding of this study was that, over the 24 month period, there was no statistically significant difference in couple scores between couples who had decided to undergo predictive testing and couples who had decided not to. The key conclusion was that predictive testing has few negative effects on couple relationships. As the authors noted, this conclusion matches the findings of several other studies (Tibben et al., 1993a; Cordori and Brandt, 1994; Quaid and Wesson, 1995; Taylor and Myers, 1997. For a look at these studies, please see “For Further Reading” at the end of this chapter).
An additional finding from the 2004 study is interesting. The couples that underwent predictive testing were categorized into couples in which the at-risk partner was a carrier and couples in which the at-risk partner was a non-carrier. Unexpectedly, the carrier couples had higher couple scores (stronger couple relationships) of statistical significance than the non-carrier couples. This suggests that, for some couples, the knowledge that their at-risk partner did not have HD had a greater negative effect on their marital relationship than the knowledge that their partner did have HD. The authors give a possible explanation: “The threat of HD may have served as a factor in the continuance of the relationship. Once this threat is removed, partners may no longer feel a duty or need to remain in the marriage to care or to be cared for.”
Another possible explanation is provided by examining family patterns via family systems theory rather than individual behavior. Family systems theory suggests that the couple relationship can be negatively affected when one or both partners have different expectations for the predictive test’s results. When the results prove to be different from expectations, conflict can arise contributing to relationship deterioration and lower couple scores. Studies by Huggins et al. and Soldan et al. have found that professional genetic counseling can benefit the couple relationship by helping partners discuss their expectations of the predictive test’s results and their coping strategies (See “Further Reading” below for links to these two studies).
What does the medical literature say about the pros and cons of predictive testing for couple relationships, especially psychosocial aspects?
Similar to the work of Richards and Williams reviewed above, a study by Decruyenaere in 2004 also used the Dyadic Adjustment Scale to measure changes in the couple relationship for 5 years following predictive testing. But the study also collected qualitative data from separate interviews with the at-risk persons and their partners. Qualitative data are useful because they can provide more thorough explanations for trends observed in couple relationship over time. The specific couple relationship examined in the Decruyenaere study was marriage.
In this study, all at-risk persons were undergoing predictive testing, with 26 carriers and 14 of their partners, and 33 of non-carriers and 17 of their partners participating in the study. The main finding was that the majority (70%) of the tested persons did not have a change in marital status over the 5 years of the study. As for the quality of the marital relationship, half of the couples reported no change in that interval compared to the quality before the predictive testing. Out of those that did report change, non-carrier couples cited less distress and more communication. Carrier couples that experienced increased relationship quality over the five years cited more mutual support.

A conclusion that can be drawn from this study is that the test result does not by itself predict outcomes in the couple relationship; even couples with negative test results for HD may experience post-test psychosocial distress and couple relationship breakdown. The important factor for couples undergoing predictive testing is whether the test result causes role shifts that upset the balance of the pre-test couple relationship. For example, two couples that received positive test results reported frustration as the partners shifted toward caretaking roles even before the people with HD showed any symptoms. In another couple tested, a woman believed to be at risk for HD gained self-esteem from a negative result. With low self-esteem before the test, she had married someone who did not match her ideals in a spouse. After the testing showed she did not have HD, she regretted her decision to marry her husband, clearly leading to relationship deterioration.
Since undesired shifts in roles may contribute to couple relationship breakdown whether the test result is positive or negative, the researchers of this study strongly support post-test counseling. Post-test counseling can help couples find and maintain a new balance that is satisfying to both partners. This counseling should include open communication between the partners, with special attention paid to the desires and worries of each partner.
Conclusions
It is clear from these studies that the psychosocial impact of predictive testing on the couple relationship is complex, with a number of factors that contribute to both positive and negative outcomes. First, the Richards and Williams study shows that pre-test discussion by the couple can be very helpful to their relationship. Such discussion can better prepare the couple for the test result by encouraging understanding of each other’s expectations of and reactions to the test result. In particular, this pre-test assessment can help identify particular challenges that the couple may face after the testing and may lead to re-consideration of testing in the first place. Complementing the Richards and Williams study, the Decruyenaere study shows the importance of post-test counseling. Post-test counseling can help protect against adverse effects of predictive testing by encouraging open discussion of each partner’s concerns as well as identification of any potential role-shifts that may disrupt the couple relationship.
Further Reading
- Decruyenaere M, Evers-Kiebooms G, Cloostermans T, Boogaerts A, Demyttenaere K, Dom R, Fryns JP. Predictive testing for Huntington’s disease: relationship with partners after testing. Clinical Genetics. 2004 Jan;65(1):24-31.
This study is not only easy-to-read but also optimistic in its finding that most marital relationships remained the same five years after predictive testing, regardless of the test results.
- Evers-Kiebooms G, Swerts A, Cassiman JJ, Van den Berghe H. The motivation of at-risk individuals and their partners in deciding for or against predictive testing for Huntington’s disease . Clinical Genetics. 1989 Jan;35(1):29-40.
This early study found that the majority of at-risk persons and their partners looked favorably upon predictive testing, although the at-risk individual and his/her partner’s reasons for deciding to take the test varied. This study took place before predictive testing began in 1993; however, the couples’ explanations for deciding on predictive testing are still eye-opening and relevant.
- Huggins et al. Predictive testing for Huntington disease in Canada : Adverse effects and unexpected results in those receiving a decreased risk . 1992 Am J Med Genet 42:508-515.
- Richards F, and Williams K. Impact on couple relationships of predictive testing for Huntington disease: a longitudinal study. American Journal of Medical Genetics Part A. 2004 Apr 15;126(2):161-9.
This is an easy-to-read article that is especially interesting because of its discussion on the benefits of pre- and post-test counseling.
- Soldan et al. Psychological model for presymptomatic test interviews: Lessons learned from Huntington disease . 2000 J Genet Couns 9:15-31.
Studies, in addition to Richards and Williams 2004, that found few negative effects of predictive testing on couple relationships:
- Codori AM, et al. Psychological costs and benefits of predictive testing for Huntington’s disease. 1994 Am J Med Genet 54:174-184.
- Quaid KA, et al. Exploration of the effects of predictive testing for Huntington disease on intimate relationships. 1995 Am J Med Genet 57:46-51.
- Taylor CA, et al. Long-term impact of Huntington disease linkage testing . 1997 Am J Med Genet 70:365-370.
- Tibben A, et al. On attitudes and appreciation 6 months after predictive DNA testing for Huntington disease in the Dutch program . 1993 Am J Med Genet 48:103-111.
-C. A. Chen 5-7-07
More


















{kind=link}
{kind=link}